
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuzuki–Miyaura coupling of arylboronic acids to gold(III)†
Ayan
Maity
a,
Amanda N.
Sulicz
a,
Nihal
Deligonul
a,
Matthias
Zeller
b,
Allen D.
Hunter
b and
Thomas G.
Gray
*a
aDepartment of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA. E-mail: tgray@case.edu
bDepartment of Chemistry, Youngstown State University, 1 University Plaza, Youngstown, Ohio 44555, USA
First published on 6th November 2014
Abstract
Gold(III) is prominent in catalysis, but its organometallic chemistry continues to be restricted by synthesis. Metal–carbon bond formation often relies on organometallic complexes of electropositive elements, including lithium and magnesium. The redox potential of gold(III) interferes with reactions of these classic reagents. Resort to toxic metals is common, including reagents based on mercury and thallium. We report that the palladium-catalyzed Suzuki–Miyaura coupling of arylboronic acids extends to cyclometalated gold(III) chlorides. Both monoarylation and diarylation are achieved. We propose a mechanism where oxidative addition to palladium with rearrangement at gold(III) fixes the stereochemistry of monoarylated intermediates. Singly arylated species form as thermodynamic isomers. These entities then go on to form diarylated complexes. Reactions proceed at room temperature, and the products are stable to air, moisture, and chromatography.
Introduction
Organometallic gold chemistry continues its broad advance, and gold(I) catalysis draws preponderant attention.1–3 Gold(I) is typically redox-neutral. Its catalytic pathways differ fundamentally from those of other transition elements, where sequences of oxidative addition and reductive elimination predominate. Gold is often called the relativistic element. Its heavy-atom character4–7 transforms the excited-state properties of luminescent gold species.8–13 Relativistic effects may also modulate the thermal reactivity of gold; this remains a topic of active inquiry.14 Gold(III) is less investigated, and is fast gaining scrutiny.15–19Contemporary studies find that gold(III) aryl complexes are luminescent, with excited states that are often ligand-localized.20 Limited syntheses hinder the emergence of organogold(III) chemistry. The major reactions that afford gold(III) complexes having three Au–C σ-bonds are transmetalation reactions from Grignard,21 organolithium,22,23 tin,24–26 or mercury(II) reagents,27 and oxidative aryl transfer from thallium(III) to gold(I).28 All of these reagents are hazardous, organolithium and magnesium reagents being pyrophoric, and tin, mercury, thallium complexes being toxic. Substrate scopes are narrow. Gold(III) is oxidizing: E°(AuIII/Au0) = +1.51 V in aqueous acid.29 Its redox character interferes in reactions with formal carbanion sources. There is a clear need for wider-ranging nonredox transformations that yield organogold(III) species.
Gold–carbon σ-bonds are covalent and modestly polar. The Pauling electronegativities of gold (2.5) and carbon (2.55) are nearly equal. We therefore hypothesized that catalytic carbon–carbon bond-forming reactions might extend to carbon–gold bond formation. Among the most powerful are cross-coupling protocols, such as the Suzuki–Miyaura coupling of organoboron species with organic halides and pseudohalides.30–40 This reaction is palladium-catalyzed, and a supporting base is normally required. Remarkably, cross-coupling reactions where the desired product is an organometallic complex are little explored.41–44 Hence, gold(III)–carbon bond formation under palladium catalysis was sought.
Results and discussion
Gold(III) dichloro complex 1 [(tpy)AuCl2] was chosen as representative. No reaction is observed between 1 and boronic acids without additives. Reactions were screened with a variety of palladium sources, phosphine ligands, and supporting bases; diaryl complexes are targeted. Table 1 summarizes outcomes. All reactions proceeded at room temperature, in contrast to the high-temperature (150 °C) transmelatations of Nevado and coworkers.45 They contrast also with boron transmetalation to gold(I), for which palladium additives are needless.46–49 We note that Mankad and Toste have reported carbon–carbon coupling in yields ranging from 0–68% in reactions of fluorogold(III) species with arylboronic acids.50 Reaction between 1 and p-fluorophenylboronic acid in the presence of Pd(PPh3)4 and Cs2CO3 in toluene led, after 24 h, to a mixture of mono- (60%) and di(p-fluorophenyl) complexes (30%), as estimated by 19F NMR spectroscopy, Table 1, entry 1. The diaryl analogue is the exclusive product after 48 h, and shows 19F singlets at δ −120.4 and −119.4 ppm, Table 1, entry 2. A similar reaction using K2CO3 as base led to 51% monoaryl product, Table 1, entry 3. The monoaryl product is recovered in 90% yield without added ligand, showing that L is not obligatory for the first coupling; Table 1, entry 4. A reaction with Pd(OAc)2, tri-(t-butylphosphine), and K2CO3 gave mono- and diaryl products in 40% and 60% yields, respectively, Table 1, entry 6. Changing the supporting base to K3PO4 afforded diaryl product in 88% yield after 16 h; no monoaryl product was observed; Table 1, entry 15.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand (L) | Base | Time (h) | Yieldb (%) | |
| Mono | Di | |||||
| a Conditions: experiments were performed with 1 (0.05 mmol), 2 (0.125 mmol), Pd catalyst (0.002 mmol), ligand (0.007 mmol), base (0.2 mmol), toluene (5 mL), rt. b Yields are based on 19F NMR relative to C6H5F as an internal standard. c Xphos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl; dppf = 1,1′-bis(diphenylphosphino)ferrocene; dcpe = 1,2-bis(dicyclohexylphosphino)ethane. | ||||||
| 1 | Pd(PPh3)4 | None | Cs2CO3 | 24 | 60 | 30 |
| 2 | Pd(PPh3)4 | None | Cs2CO3 | 48 | 0 | 85 |
| 3 | Pd(PPh3)4 | None | K2CO3 | 16 | 51 | 0 |
| 4 | Pd(OAc)2 | None | K2CO3 | 16 | 90 | 0 |
| 5 | Pd(OAc)2 | PPh3 | K2CO3 | 16 | 38 | 0 |
| 6 | Pd(OAc)2 | PtBu3 | K2CO3 | 16 | 40 | 60 |
| 7 | Pd2dba3 | Xphosc | K2CO3 | 16 | 50 | 44 |
| 8 | Pd(dppf)Cl2 | dppfc | K2CO3 | 16 | 27 | 0 |
| 9 | Pd(OAc)2 | dcpec | K2CO3 | 16 | 34 | 0 |
| 10 | PdCl2 | PtBu3 | K2CO3 | 16 | 31 | 0 |
| 11 | Pd(OAc)2 | PCy3 | K2CO3 | 16 | 49 | 0 |
| 12 | Pd(OAc)2 | PtBu3 | NaOMe | 16 | 12 | 47 |
| 13 | Pd(OAc)2 | PtBu3 | KOH | 16 | 25 | 25 |
| 14 | Pd(OAc)2 | PtBu3 | n Bu4NF | 16 | 0 | 0 |
| 15 | Pd(OAc)2 | PtBu3 | K3PO4 | 16 | 0 | 88 |
| 16 | Pd2dba3 | PCy3 | K3PO4 | 16 | 5 | 86 |
| 17 | Pd(OAc)2 | [HP(tBu)3]BF4 | K3PO4 | 16 | 0 | 90 |
| 18 | None | [HP(tBu)3]BF4 | K3PO4 | 16 | 0 | 0 |
Use of the air- and moisture-stable phosphonium salt [HP(tBu)3]BF4 gave the diarylated gold(III) complex in unimpaired yields, Table 1 entry 17; and allowed reaction components to be weighed in open air. (However, reactions proceeded under an argon atmosphere.) Use of the stronger bases NaOMe and KOH gave diminished yields of both products, Table 1 entries 12 and 13. No reaction was observed in the presence of anhydrous nBu4NF, Table 1 entry 14.
The reaction accelerated with addition of 2-propanol. With K2CO3 as supporting base, the reaction was complete in 10 h, Table 2 entry 10. With K3PO4, the reaction was completed in 4 h, Table 2, entry 9. The reaction conditions described in Table 2, entry 10, involving K2CO3 were chosen for subsequent work for its higher yield across a variety of boronic acid substrates.
| Entry | Base | Solvent | Time (h) | Yieldb (%) | |
|---|---|---|---|---|---|
| Mono | Di | ||||
| a Conditions: experiments were performed with 1 (0.05 mmol), 2 (0.125 mmol), Pd(OAc)2 (0.002 mmol), [HP(tBu)3]BF4 (0.007 mmol), base (0.2 mmol), indicated solvent-solvent mixture (5 mL), rt. b Yields are based on 19F NMR relative to C6H5F as internal standard. | |||||
| 1 | K3PO4 | 1,4-Dioxane | 16 | 0 | 0 |
| 2 | K3PO4 | DMF | 16 | 0 | 0 |
| 3 | K3PO4 | 1% iPrOH in toluene | 16 | 10 | 70 |
| 4 | K3PO4 | 1% EtOH in toluene | 16 | 30 | 60 |
| 5 | K3PO4 | THF | 16 | 50 | 50 |
| 6 | Li3PO4 | Toluene | 16 | 0 | 0 |
| 7 | KOH | Toluene | 16 | 30 | 10 |
| 8 | K2CO3 | 10% H2O in THF | 16 | 0 | 0 |
| 9 | K3PO4 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene–iPrOH :1 toluene–iPrOH |
4 | 0 | 82 |
| 10 | K2CO3 | 1:1 toluene–iPrOH |
10 | 0 | 85 |
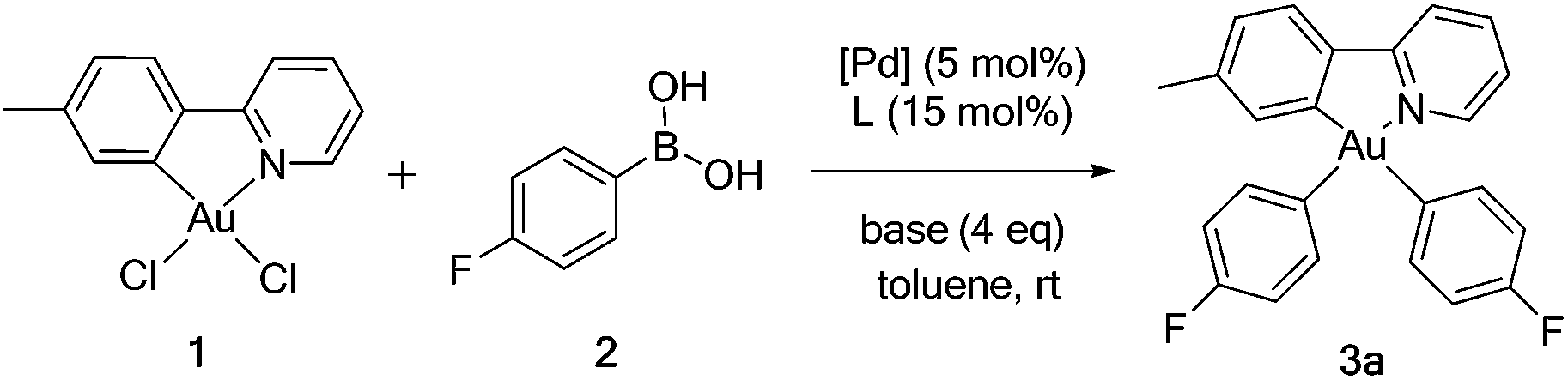
The standardized protocol was applied to the synthesis of a range of gold(III) aryls, Table 3. Aryl groups with electron-withdrawing (3a–f), electron-neutral (3g–l) and electron-releasing substituents (3m–o) were bound to gold. Isolated yields range from 42–78%. Efficiencies are comparable for boronic acids with electron-withdrawing or releasing substituents. Organogold complexes are readily prepared having oxidized substituents that, like gold(III) itself, degrade on treatment with lithium reagents or other formal carbanion sources. The products are purified by column chromatography on basic alumina. They are stable as solids to air and water.
|
The reaction is specific for auration at borylated carbons. Complexes 3a, 3c, 3d, 3j, 3l, and 3n were characterized by X-ray diffraction crystallography. Thermal ellipsoid diagrams are deposited as ESI.† In each structure, p-tolylpyridyl chelates as a bidentate ligand. Geometric parameters about the metal are within ranges typical of Au(III).51Trans-influences of carbon and nitrogen are evident in gold–aryl carbon bond distances. In all instances, the Au–C bond trans to carbon is significantly longer52 than that trans to nitrogen.
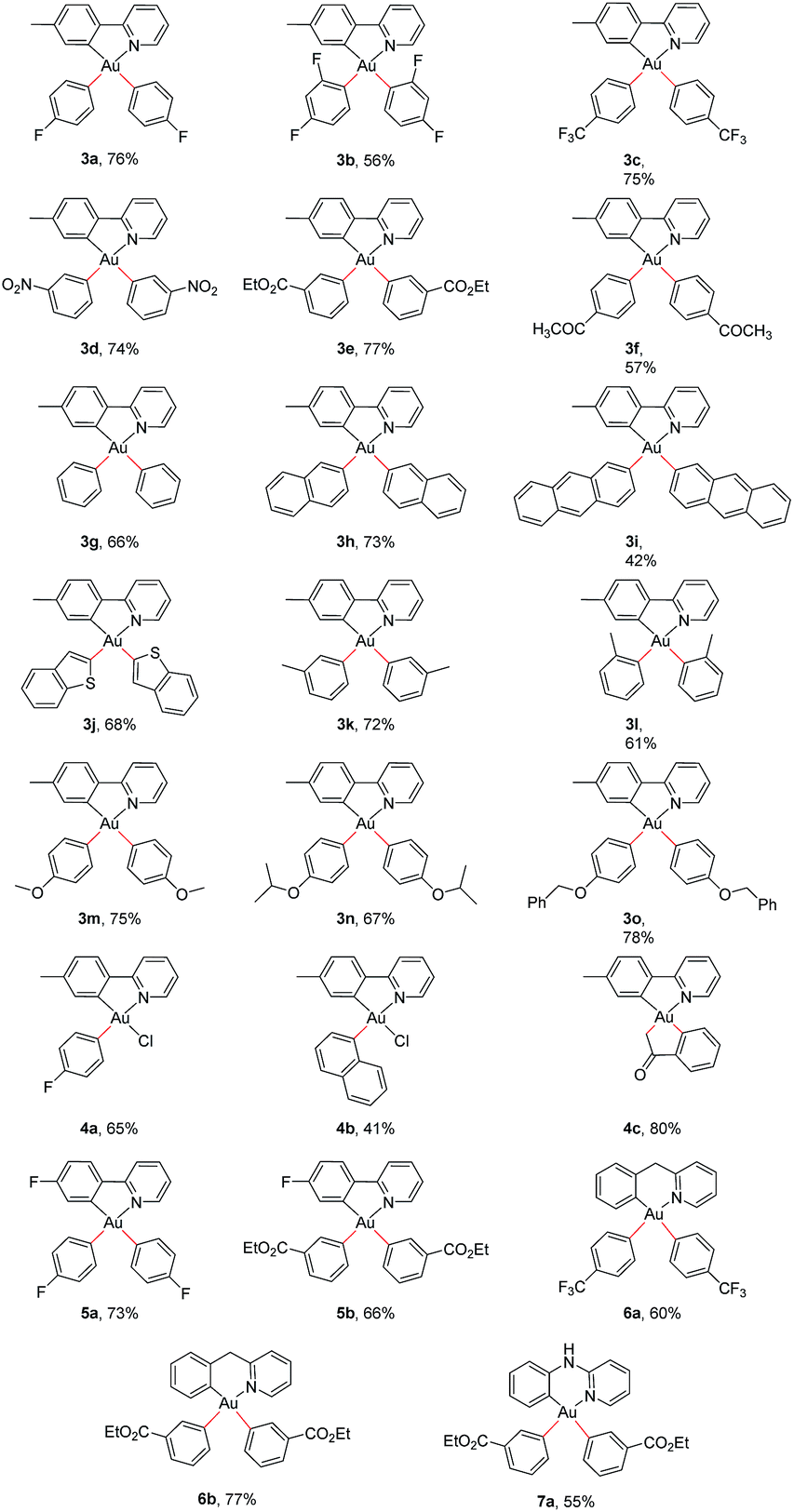
While surveying reaction parameters for diarylation, monoarylated products were isolated. Complexes 4a and 4b were isolated as products of incomplete aryl transfer. 1H NMR experiments show that both products form as a single isomer. The crystal structure of 4a appears as Fig. 1(a); that of 4b is provided as ESI.† The p-fluorophenyl ligand binds trans to the pyridyl nitrogen despite the kinetic trans effect. Nevertheless, the observed structure of 4a is expected to be more stable than diastereomer 4a′ where p-fluorophenyl binds opposite carbon, Fig. 2.
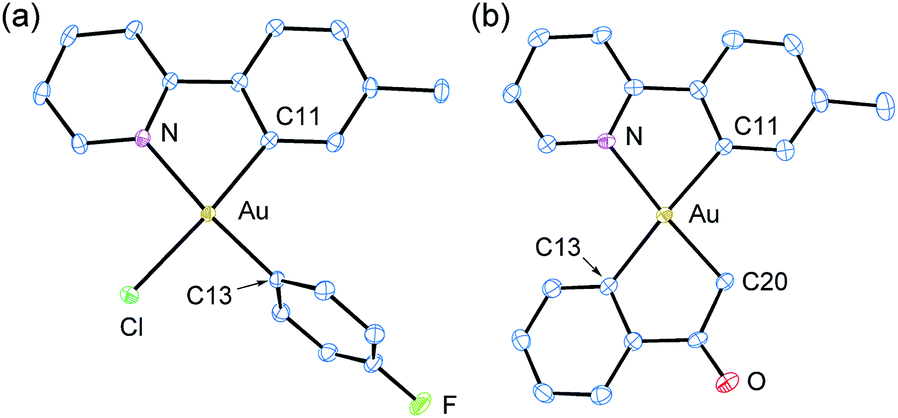 | ||
| Fig. 1 (a) Crystal structure of p-fluorophenyl complex 4a. Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Unlabeled atoms are carbon. Selected interatomic distances (Å): C11–Au, 2.023(4); Au–N, 2.116(3); C13–Au, 2.018(4); Au–Cl, 2.3707(9). Selected angles (°): N–Au–C11, 81.05(14); C13–Au–Cl: 91.07(10). (b) Crystal structure of C-enolate 4c. Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Unlabeled atoms are carbon. Selected interatomic distances (Å): C11–Au, 2.058(5); Au–N, 2.134(4); C13–Au, 2.080(5); C20–Au, 2.041(6). Selected angles (°): N–Au–C11, 79.62(18); C13–Au–C20, 80.8(2). | ||
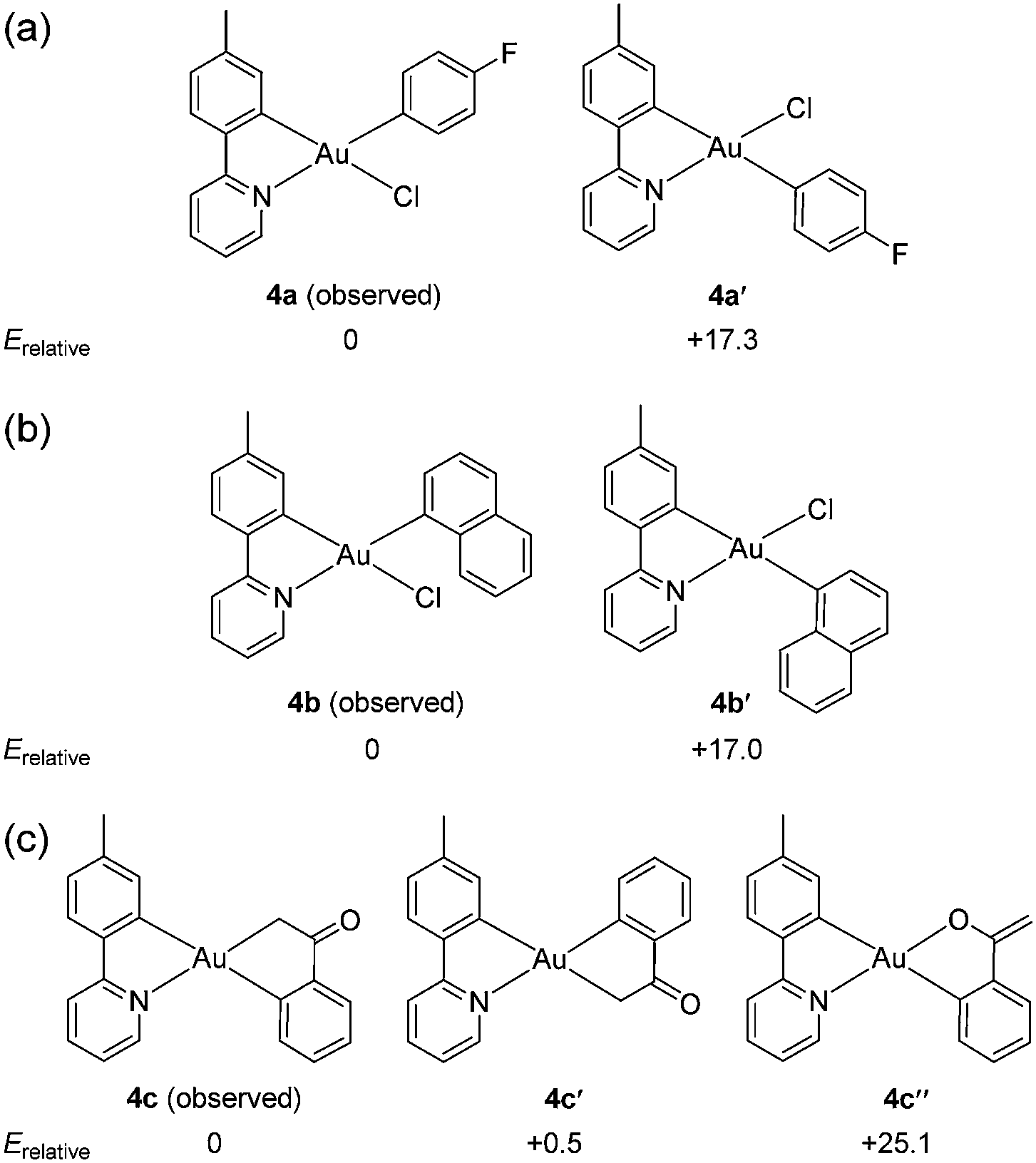 | ||
| Fig. 2 Relative energies in kcal mol−1 calculated for (a) mono-p-fluorophenyl isomers 4a and 4a′; (b) mono-1-naphthyl isomers 4b and 4b′; and (c) enolate complexes 4c, 4c′, and 4c′′. Energies are calculated from sums of electronic and thermal free energies. | ||
Density-functional theory (DFT) calculations using the parameter-free hybrid functional of Perdew, Burke, and Ernzerhof53 were used to evaluate the relative thermochemistry of isomers. The calculations indicate that 4a is 17.3 kcal mol−1 more stable than 4a′ with trans-disposed carbons, Fig. 2. Results are similar for 1-naphthyl complex 4b (Table S1, ESI†). Hence, binding of the first aryl ligand selects for the thermodynamic product.
The stereochemistry of 4a and 4b is surprising in that the aryl ligand is trans to nitrogen. If arylation proceeds in a single step, then the shorter Au–Cl bond ostensibly breaks first. Attempts to grow single crystals of 1 failed. However, the structure of the related complex dichloro(2-(4-fluorophenyl)pyridine)gold(III) was obtained. This complex differs from 1 in that fluorine substitutes for methyl in the C^N ligand. The structure (ESI†) shows a longer Au–Cl bond trans to carbon. Pertinent interatomic distances are 2.3750(15) Å for Au–Cl trans to C and 2.2721(17) Å for Au–Cl trans to N. Thus, the shorter Au–Cl bond opposite nitrogen is sacrificed in the first arylation.
Geometry optimization of [(tpy)AuIIICl]+ fragments tells against a limiting dissociative mechanism initiated by chloride loss. Energy minimization of [(tpy)AuIIICl]+ leads to a T-shaped structure where the space trans to carbon is empty. An incoming ligand is expected to attack this open site, leading to a stereochemistry unlike that observed. The aryl ligands in 4a and 4b are opposite nitrogen. We therefore propose transmetalation by an associative or associative interchange mechanism, possibly mediated by palladium, that avoids mutually trans carbon atoms and leads to the thermodynamic isomers 4a and 4b.
Reactions of 1 with (2-acetylphenyl)boronic acid yielded the singly arylated 4c. 1H and 13C NMR spectra indicate a single species in solution. This complex is a C-bound enolate stabilized by chelation of the benzene ring. The pKa of acetophenone is 24.7 in dimethyl sulfoxide.54 Deprotonation of the α-carbon atom may simply result from the action of base. Vapor diffusion of pentane into dichloromethane solution afforded diffraction-quality crystals. The structure of 4c appears in Fig. 1(b). The sp3-hybridized carbon atom lies trans to the pyridyl nitrogen of the tpy ligand. A ν(CO) stretching frequency at 1666 cm−1 and a 13C{1H} NMR resonance at δ 209 ppm both indicate retention of the C-bound enolate geometry. It is noteworthy that gold(III) binds to the softer carbon, rather than oxygen, as is common for oxidized metals of the earlier d-block.55,56
DFT calculations find that the C-bound enolate is some 25.1 kcal mol−1 more stable than the O-bound tautomer. Observed 4c is virtually isoenergetic with the –C-bound “flipped” enolate where sp3-hybridized carbon binds opposite the tpy tolyl carbon. Line drawings and relative energies appear as in Fig. 2(c). Experimentally, a single complex is recovered, not a mixture, suggesting that the reaction is specific for 4c.
A potential mechanism for the formation of 3a–o appears in Scheme 1. Oxidative addition of 1 to palladium(0) presumably occurs at the longer Au–Cl bond trans to carbon. Rearrangement follows, possibly through a five-coordinate intermediate that pseudorotates.57,58 Formation of a palladium alkoxide or hydroxide intermediate precedes transmetalation from boron, in keeping with results from Hartwig, Amatore, Jutand, and their respective co-workers.59–61 Reductive elimination62 yields monosubstituted products with the stereochemistry established for 4a and 4b. Singly arylated products re-enter the catalytic process, undergo transmetalation and reductive elimination, and emerge as diaryls. Experiments that test this hypothesis are underway.
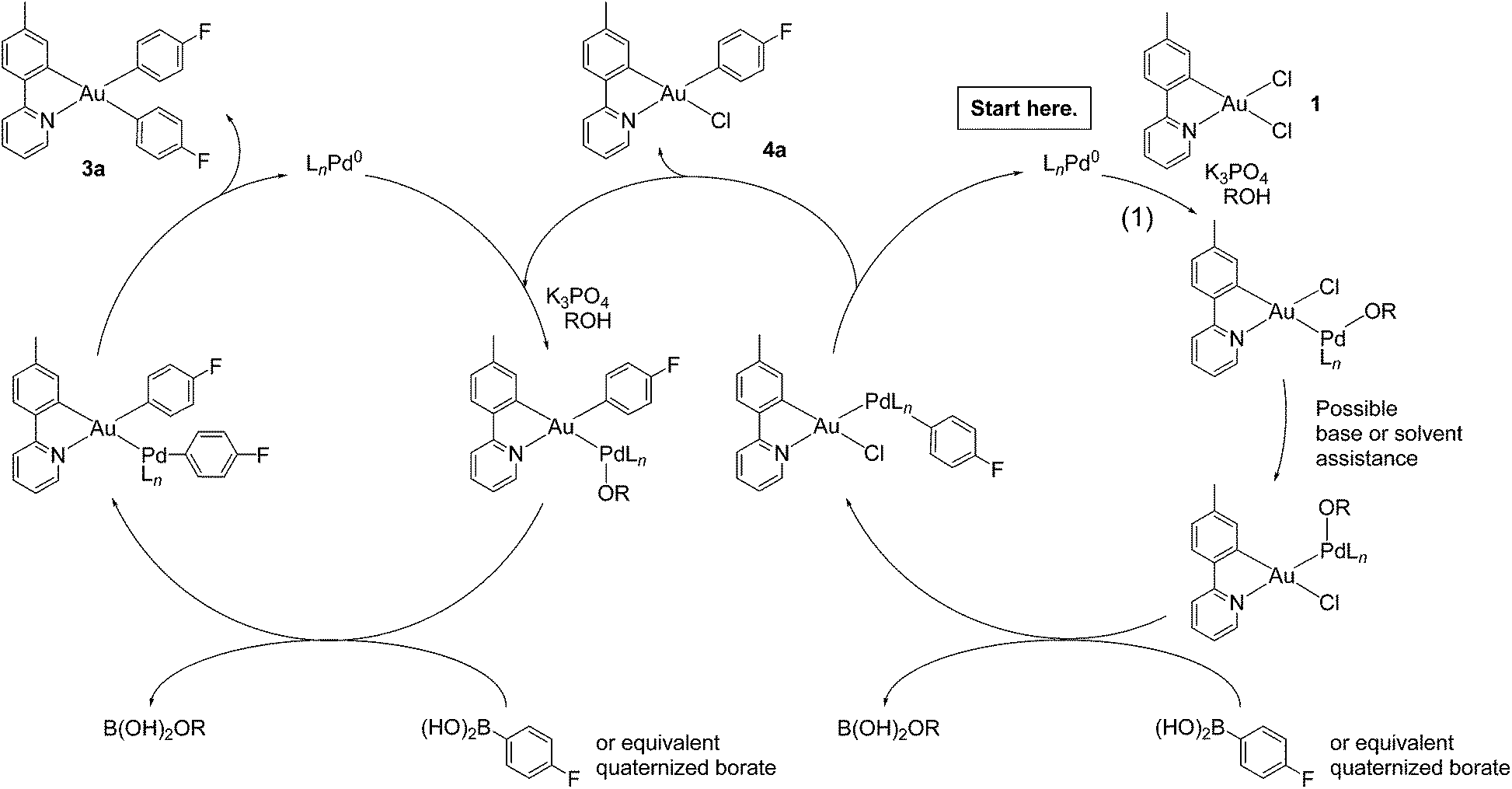 | ||
| Scheme 1 Proposed mechanism of gold(III) monoarylation (right cycle) and subsequent diarylation (left cycle). | ||
Conclusions
We report catalytic arylation of gold(III) through Suzuki–Miyaura couplings at room-temperature. The reaction is palladium-mediated and requires an assisting base; the electrophilic reacting partner is a cyclometalated gold(III) dichloro complex. Screening experiments show that palladium(II) acetate is an effective catalyst precursor when combined with potassium phosphate and tri-(t-butylphosphine). The phosphine is conveniently delivered as an air- and moisture-stable phosphonium tetrafluoroborate salt. Variously substituted arylboronic acids couple to gold in similar yields. An ortho-substituted enolizable ketone leads to a C-bound enolate, without continuing to form a diaryl. Spectroscopic characterization of the enolate complex indicates that a single product forms. Crystal-structure determination shows that the sp3-hybridized carbon binds opposite the pyridyl nitrogen, and a phenyl carbon binds trans to the tolyl carbon of the C^N ligand. DFT calculations find the C-enolate to be more stable than an O-bound tautomer.Compounds 4a and 4b are singly arylated products that were characterized structurally. Spectral data indicate a single isomer of each in solution. Both crystal structures show aryl substitution trans to the C^N nitrogen atom. This stereospecificity is counterintuitive given the trans-influence of carbon: the shorter Au–Cl bond disappears first. We propose that oxidative addition of an Au–Cl bond to palladium happens trans to the tolyl carbon of the C^N ligand. Rearrangement yields the more stable isomer, with a gold–carbon bond trans to nitrogen. Reductive elimination generates monoaryls of the observed stereochemistry and liberates palladium. The monoaryl product, if not isolated, can then re-enter the catalytic sequence to yield diaryls. The photophysical properties of gold(III) aryls are being investigated, as are extensions to other gold complexes and to other metals.
Experimental
Synthesis of [(tpy)Au(p-C6H5F)2] (3a)
In a 100 mL Schlenk flask, Pd(OAc)2 (2.6 mg, 0.011 mmol) was dissolved in 10 mL dry toluene under argon. [HP(tBu)3]BF4 (9.9 mg, 0.034 mmol) and K2CO3 (128 mg, 0.917 mmol) were added, resulting in a pale yellow solution that was allowed to stir for 5 min at room temperature under argon. To this solution was added 1 (100 mg, 0.229 mmol) followed by 4-fluorophenylboronic acid (80.2 mg, 0.573 mmol) and 10 mL dry 2-propanol. The reaction mixture was degassed in three freeze-pump-thaw cycles. Finally the reaction mixture was sealed and left stirring for 10 h at room temperature. Reaction progress was monitored by TLC. After the reaction was complete, the volatiles were removed under reduced pressure. The resulting crude product was re-dissolved in 10 mL methylene chloride and was passed through a plug of Celite. The volume of solvent was reduced, and the crude mixture was then purified by a short basic-alumina column using variant polarity between hexanes and hexanes–diethyl ether (1:3, v/v). The desired product was eluted using hexanes–diethyl ether (1:2, v/v). Removal of solvent left a white solid, which was dried under vacuum for 6 h. Yield 96 mg (76%); TLC (hexanes–diethyl ether, 40:60 v/v): Rf = 0.68; 1H NMR (400 MHz, CD2Cl2): δ 8.06 (dt, J = 5.9, 1.0 Hz, 1H), 7.95 (d, J = 4.7 Hz, 2H), 7.71 (d, J = 8.1 Hz, 1H), 7.45 (d, J = 6.7 Hz, 1H), 7.43 (d, J = 6.5 Hz, 1H), 7.40 (d, J = 6.1 Hz, 1H), 7.37 (d, J = 6.1 Hz, 1H), 7.18 (q, J = 2.6 Hz, 1H), 7.09 (dd, J = 8.1, 1.2 Hz, 1H), 7.02–6.90 (m, 4H), 6.78 (d, J = 1.1 Hz, 1H), 2.22 (s, 3H); 19F NMR (376.1 MHz, CD2Cl2): δ −119.4 (m, 1F), −120.4 (m, 1F); UV/Vis (methylene chloride): λmax, nm (ε, M−1 cm−1) 272 (sh, 31000), 329 (12000); emission (methylene chloride): λem, nm (int.) 467 (170), 494 (199); analysis (calcd, found for C24H18AuF2N): C (51.90, 52.21), H (3.27, 3.31), N (2.52, 2.88).
Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering, under Award DE-FG02-13ER46977 to T. G. G. X-ray diffractometers at Youngstown State University were funded by NSF Grants 0087210 and 1337296, Ohio Board of Regents Grant CAP-491, and by Youngstown State University.Notes and references
- A. S. K. Hashmi and F. D. Toste, in Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS.
- H. G. Raubenheimer and H. Schmidbaur, J. Chem. Educ. DOI:10.1021/ed400782p.
- P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113–4130 CrossRef PubMed.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- P. Schwerdtfeger, P. D. W. Boyd, S. Brienne and A. K. Burrell, Inorg. Chem., 1992, 31, 3411–3422 CrossRef CAS.
- N. Kaltsoyannis, Dalton Trans., 1997, 1–11 RSC.
- D. V. Partyka, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2007, 26, 3279–3282 CrossRef CAS.
- L. Gao, M. A. Peay, D. V. Partyka, J. B. Updegraff III, T. S. Teets, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2009, 28, 5669–5681 CrossRef CAS.
- R. A. Vogt, M. A. Peay, T. G. Gray and C. E. Crespo-Hernández, J. Phys. Chem. Lett., 2010, 1, 1205–1211 CrossRef CAS.
- R. A. Vogt, T. G. Gray and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2012, 134, 14808–14817 CrossRef CAS PubMed.
- V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- C. Yang, M. Messerschmidt, P. Coppens and M. A. Omary, Inorg. Chem., 2006, 45, 6592–6594 CrossRef CAS PubMed.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7411–7420 CrossRef CAS PubMed.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS PubMed.
- M. J. Ghidiu, A. J. Pistner, G. P. A. Yap, D. A. Lutterman and J. Rosenthal, Organometallics, 2013, 32, 5026–5029 CrossRef CAS.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778–1781 CrossRef CAS PubMed.
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777–7782 CrossRef CAS PubMed.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC.
- S. Komiya and A. Shibue, Organometallics, 1985, 4, 684–687 CrossRef CAS.
- A. Szentkuti, M. Bachmann, J. A. Garg, O. Blacque and K. Venkatesan, Chem.–Eur. J., 2014, 20, 2585–2596 CrossRef CAS PubMed.
- J. A. Garg, O. Blacque, T. Fox and K. Venkatesan, Inorg. Chem., 2010, 49, 11463–11472 CrossRef CAS PubMed.
- R. Usón, J. Vicente, J. A. Cirac and M. T. Chicote, J. Organomet. Chem., 1980, 198, 105–112 CrossRef.
- B. David, U. Monkowius, J. Rust, C. W. Lehmann, L. Hyzak and F. Mohr, Dalton Trans., 2014, 43, 11059–11066 RSC.
- T. N. Zehnder, O. Blacque and K. Venkatesan, Dalton Trans., 2014, 43, 11959–11972 RSC.
- K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350–4365 CrossRef CAS PubMed.
- R. Uson, A. Laguna, M. Laguna, E. Fernandez, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1982, 1971–1976 RSC.
- Inorg. Chim. Acta, Technical note, 1995, 239, 189 DOI:10.1016/0020-1693(95)90093-4.
- J. C. H. Lee and D. G. Hall, in Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, Germany, 2014, vol. 1, pp. 65–132 Search PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- C. Valente and M. G. Organ, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine, and Materials, Wiley-VCH, Weinheim, Germany, 2011, vol. 1, pp. 213–262 Search PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed.
- F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2008, 64, 3047–3101 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- A. Suzuki, Suzuki Coupling, Organic Syntheses via Boranes, Aldrich, Milwaukee, 2003, vol. 3 Search PubMed.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 36, 3437–3440 CrossRef.
- C. Lo Sterzo, M. M. Miller and J. K. Stille, Organometallics, 1989, 8, 2331–2337 CrossRef CAS.
- C. Lo Sterzo and J. K. Stille, Organometallics, 1990, 9, 687–694 CrossRef CAS.
- C. Lo Sterzo, Organometallics, 1990, 9, 3185–3188 CrossRef CAS.
- A. Ricci, F. Angelucci, M. Bassetti and C. Lo Sterzo, J. Am. Chem. Soc., 2002, 124, 1060–1071 CrossRef CAS PubMed.
- M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328–1332 CrossRef CAS.
- D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188–8191 CrossRef CAS PubMed.
- J. E. Heckler, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2012, 51, 5924–5928 CrossRef CAS PubMed.
- D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Inorg. Chem., 2012, 51, 8394–8401 CrossRef CAS PubMed.
- H. K. Lenker, T. G. Gray and R. A. Stockland, Jr., Dalton Trans., 2012, 41, 13274–13276 RSC.
- N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859–12861 CrossRef CAS PubMed.
- H. Schmidbaur, A. Grohmann and M. E. Olmos, in Gold: Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, Wiley, Chichester, 1999, pp. 647–746 Search PubMed.
- G. H. Stout and L. H. Jensen, X-ray Structure Determination: A Practical Guide, Wiley Interscience, 2nd edn, 1989; pp. 404–405 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- J. Cámpora, C. M. Maya, P. Palma, E. Carmona, E. Gutiérrez-Puebla and C. Ruiz, J. Am. Chem. Soc., 2003, 125, 1482–1483 CrossRef PubMed.
- H. S. Soo, P. L. Diaconescu and C. C. Cummins, Organometallics, 2004, 23, 498–503 CrossRef CAS.
- A. L. Casado and P. Espinet, Organometallics, 1998, 17, 954–959 CrossRef CAS.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed.
- C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CrossRef CAS PubMed.
- C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159–164 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. CCDC [1009034–1009043]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02148g |
| This journal is © The Royal Society of Chemistry 2015 |