
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiagnosing the miR-141 prostate cancer biomarker using nucleic acid-functionalized CdSe/ZnS QDs and telomerase†
Amily Fang-ju
Jou
a,
Chun-Hua
Lu
b,
Yen-Chuan
Ou
*c,
Shian-Shiang
Wang
c,
Shih-Lan
Hsu
d,
Itamar
Willner
*b and
Ja-an Annie
Ho
*a
aBioAnalytical Chemistry and Nanobiomedicine Laboratory, Department of Biochemical Science & Technology, National Taiwan University, Taipei 10617, Taiwan. E-mail: jaho@ntu.edu.tw
bInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: willnea@vms.huji.ac.il
cVACRS Division of Urology, Department of Surgery, Taichung Veterans General Hospital, Taichung 40705, Taiwan. E-mail: ycou228@gmail.com
dDepartment of Education and Research, Taichung Veterans General Hospital, Taichung 40705, Taiwan
First published on 9th September 2014
Abstract
The microRNA, miR-141, is a promising biomarker for prostate cancer. We implement here a two-step sensing platform for the sensitive detection of miR-141. The first step involves the use of semiconductor CdSe/ZnS quantum dots (QDs) modified by FRET quencher-functionalized nucleic acids, that include the recognition sequence for miR-141 and a telomerase primer sequence for the second step of the analytical platform. Subjecting the probe-modified QDs to miR-141, in the presence of duplex specific nuclease, DSN, leads to the formation of a miR-141/probe duplex and to its DSN-mediated cleavage, while regenerating the miR-141. The DSN-induced cleavage of the quencher units leads to the activation of the fluorescence of the QDs, thus allowing the optical detection of miR-141 with a sensitivity corresponding to 1.0 × 10−12 M. The nucleic acid residues associated with the QDs after cleavage of the probe nucleic acids by DSN act as primers for telomerase. The subsequent telomerase/dNTPs-stimulated elongation of the primer units forms G-quadruplex telomer chains. Incorporation of hemin in the resulting G-quadruplex telomer chains yields horseradish peroxidase-mimicking DNAzyme units, that catalyze the generation of chemiluminescence in the presence of luminol/H2O2. The resulting chemiluminescence intensities provide a readout signal for miR-141, DL = 2.8 × 10−13 M. The first step of the sensing platform is non-selective toward miR-141 and the resulting fluorescence may be considered only as an indicator for the existence of miR-141. The second step in the sensing protocol, involving telomerase, provides a selective chemiluminescence signal for the existence of miR-141. The two-step sensing platform is implemented for the analysis of miR-141 in serum samples from healthy individuals and prostate cancer carriers. Impressive discrimination between healthy individuals and prostate cancer carriers is demonstrated.
Introduction
Prostate cancer (PC) is the most frequently diagnosed cancer, and is a major cause of cancer-related mortality in males.1 The rapid, reliable and cost-effective detection of PC could have a tremendous clinical impact for diagnosis, prognosis, and treatment response. The quantitative analysis of the prostate-specific antigen (PSA) in serum is currently used for diagnosing PC and treatment response. The use of PSA as a reliable biomarker for PC has, however, been criticized, and its diagnostic value questioned.2,3 Elevated amounts of PSA are not specific to the malignant disease, and indolent prostate tumors are false-positively identified as PC. Additionally, the PSA diagnostic test gives ca. 15% false-negative results. Here we introduce a new two-step optical sensing platform for analysing the microRNA, miR-141, which is a biomarker for PC. The sensing platform is applied to the analysis of clinical samples, and it is suggested as a substitute, or complementary, test for the PSA immunoassay.Circulating miRNAs are short oligonucleotides (19–26 bases) that are suggested to be important regulators of biological functions. Up- or down-regulation of the expression of miRNAs affects cellular processes, such as proliferation or apoptosis, and links between miRNAs and cancer development exist.4,5 Accordingly, miRNAs provide a rich platform of biomarkers for different diseases. Indeed, miRNA-141 (miR-141) has been found to be up-regulated in PC specimens, and suggested as a potentially useful biomarker for PC.6
Recent research efforts are directed toward the application of nanotechnological tools and methods to develop amplified detection platforms for nucleic acids.7 Different amplified sensing platforms of nucleic acids have applied DNA machinery for the autonomous synthesis of catalytic nucleic acids (DNAzymes) as amplifying labels.8–16 Alternatively, the regeneration of the target-analyte by biocatalytic transformations, such as exonuclease III or nicking enzymes, was used as a versatile amplification path for DNA sensing events.17–20 Additionally, the unique optical properties of semiconductor quantum dots (QDs) were broadly applied as nanomaterials for developing optical sensors and biosensors.21,22 Specifically, QDs were used to develop luminescent DNA sensors using FRET,23 CRET24,25 or electron-transfer quenching26 as readout signals. The Exo III-catalyzed regeneration of the target DNA using quencher–nucleic acid-functionalized QDs was also implemented for the amplified, multiplexed analysis of DNAs.27 In the present study, we introduce a two-step miR-141 analysis scheme, using CdSe/ZnS QDs and telomere units for the fluorescent and chemiluminescent detection of miR-141.
Results and discussion
The first step in the amplified sensing platform for miR-141 involves the optical detection of miRNA using semiconductor QDs, as outlined in Fig. 1A. Water-soluble glutathione-functionalized CdSe/ZnS QDs were reacted with 5′-amino-functionalized nucleic acid (1) modified at its 3′-end with the BHQ2 quencher. The average coverage of the nucleic acid (1) on the QDs was determined spectroscopically to be 8 units per particle. The nucleic acid associated with the QDs includes domains I and II. The sequence of II is complementary to miR-141, while domain I consists of the sequence that acts as the telomerase primer (for its function vide infra). In the presence of miR-141, a duplex between miR-141 and domain II of (1) is formed. Subjecting the system to the duplex-specific nuclease, DSN, leads to the cleavage of the DNA strand associated with the duplex (1)/miR-141. This results in the removal of the BHQ-quencher, the sequential degradation of the duplex region II of (1), and the recycling of the miR-141; this binds to another probe (1) associated with the QDs. Electrophoretic measurements indicate that the DSN-mediated digestion of domain II of (1) results in a short nucleic acid single-stranded tether linked to the QDs, which is composed of domain I extended by 2–4 bases of II. This tether is essential for the secondary telomerase-stimulated detection of miR-141 (vide infra). The DSN-stimulated cleavage of the probe strands leads to the removal of the quencher units and to the switching “on” of the luminescence of the QDs. The sequential recycling of the target miR-141 by the DSN-mediated process provides an amplification path for the sensing process. Fig. 1B depicts the time-dependent luminescence changes upon subjecting the (1)-modified QDs to miR-141, 1 × 10−7 M, in the presence of DSN. The luminescence of the QDs increases with time, and it reaches a saturation value after ca. 60 min. The time-dependent fluorescence changes correspond to the dynamics of the DSN-induced cleavage of the quencher units associated with the QDs, through the regeneration of the miR-141 target. Fig. 1C shows the luminescence spectra recorded upon subjecting the (1)-modified QDs to various concentrations of miR-141, in the presence of DSN, for a fixed time-interval of one hour. As the concentration of miR-141 increases, the luminescence is intensified, consistent with the higher degree of cleaved-off quencher units. Fig. 1C, inset, shows the resulting calibration curve. The lowest measured concentration of miR-141 by this scheme corresponded to 1.0 × 10−12 M. (For optimization of other parameters of the sensing platform, see ESI Fig. S1†.)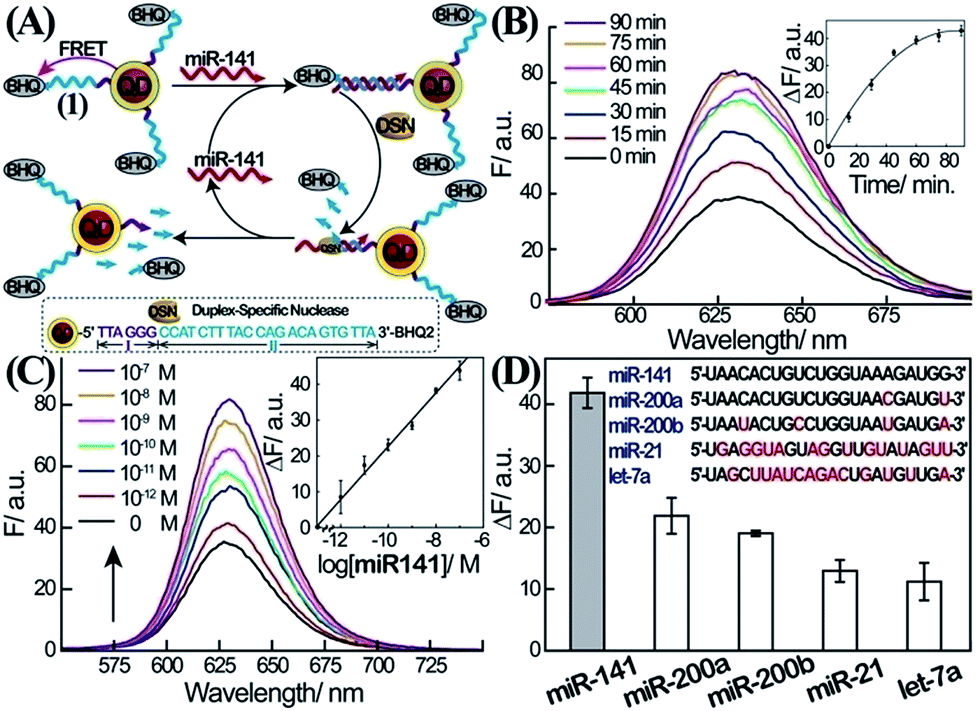 | ||
| Fig. 1 Amplified detection of miR-141 through DSN-stimulated cleavage of the miR-141 target. (A) Schematic fluorescence analysis of miR-141 by nucleic acid-functionalized CdSe/ZnS quantum dots (QDs) through DSN-stimulated regeneration of the miR-141 target. (B) Time-dependent fluorescence spectra observed upon the interaction of the (1)-modified QDs with miR-141, 1 × 10−7 M, and DSN, 0.2 U. Inset: fluorescence changes, ΔF = F − F0, at λ = 627 nm, as a function of the interaction time of the QDs with miR-141 and DSN. (C) Fluorescence spectra corresponding to the (1)-functionalized QD system upon analyzing different concentrations of miR-141 using DSN as a target regeneration biocatalyst. Fluorescence spectra were recorded after a fixed time-interval of one hour of the reaction. Inset: derived calibration curve corresponding to the resulting fluorescence changes, ΔF = F − F0, at λ = 627 nm at different concentrations of the target miR-141. (D) Fluorescence changes; ΔF = F − F0, observed upon the interaction of the (1)-functionalized QDs with different miRs, 1 × 10−7 M, and DSN, 0.2 U, for one hour. All error bars in the figures indicate standard deviations, using N = 3 experiments. | ||
The selectivity of the sensing of miR-141 is an important issue, since other miRs include homologous nucleotide domains to the miR-141, and hence might interfere with the specific analysis of miR-141. Accordingly, the fluorescence responses of the (1)-modified QDs to miR-200a, miR-200b, miR-21, and let-7a were examined (Fig. 1D). Evidently, miR-200a and 200b, exhibiting substantial sequence similarities to miR-141, reveal significant fluorescence signals (52% and 46% of the miR-141 signal, respectively); this is consistent with the fact that these miRs hybridize with (1), leading to their partial cleavage by DSN. While the relative concentrations of miR-200a and miR-200b, as compared to miR-141 in real biological samples, are unknown, their fluorescence response might, indeed, perturb the quantitative measurement of miR-141. One should note, however, that despite the base similarities, miR-200a and miR-200b include several base mismatches, and the 3′-ends of the two miRs, in particular, are non-complementary to the probe (1). As a result, the DSN-stimulated cleavage of the duplex does not yield the domain I extended by 2–4 bases which is generated in the presence of miR-141; instead domain I is extended by >8 bases (this difference is important to induce selectivity for miR-141 analysis in the second step, vide infra.)
To overcome the selectivity limitation of the optical detection of miR-141, and to amplify the primary recognition event of miR-141, we applied telomerase as a catalytic amplifier. Telomerase is a ribonucleoprotein that includes a nucleic acid template in the protein backbone.28 It is over-expressed in cancer cells, and catalyzes the extension of the telomerase primer in the presence of the dNTPs bases, to form telomere chains consisting of telomeric repeating units, TTAGGG.29 The resulting telomere chains self-assemble into G-quadruplexes, and in the presence of hemin, yield telomeric hemin/G-quadruplex horseradish peroxidase-mimicking DNAzyme units. These have been found to catalyze the oxidation of luminol by H2O2 to generate chemiluminescence.30 We have harnessed the catalytic properties of the telomeric hemin/G-quadruplex products, to amplify and enhance the selectivity of the detection of miR-141 on the QDs support. The DSN-stimulated cleavage of the duplex miR-141/domain II of (1) results in the hydrolytic digestion of domain II and the regeneration of miR-141, while the domain I tethered to the QDs is extended by 2–4 bases belonging to domain II. We found that the tether I extended by 2–4 bases, CCAT, acted as a primer for telomerase. Accordingly, the QDs functionalized with the domain I extended by 2–4 bases were treated with telomerase extracted from PC-3 cells in the presence of the dNTP mixture (Fig. 2A). This resulted in the telomerization of the single-stranded tether associated with the QDs and, in the presence of hemin, to the self-assembly of the catalytic-telomeric hemin/G-quadruplexes that generated chemiluminescence as a readout for miR-141. It was noted that the incomplete complementarity of the different miRs to the probe (1) led, in the presence of DSN, to perturbed domains I extended by overhangs of >8 bases. These extended sequences are not recognized by telomerase. Consequently, the resulting modified nucleotides linked to the QDs cannot initiate telomerization; this implies that the secondary telomerase-stimulated elongation of the tether associated with the QDs is prohibited, and selectivity to the miR-141/DSN-generated sequence is achieved. Fig. 2B shows the time-dependent chemiluminescence spectra observed upon the treatment of the (1)-functionalized QDs with a fixed concentration of miR-141 (1 × 10−7 M) and DSN, for a fixed time-interval (60 minutes), and their subsequent interaction with telomerase and dNTPs for variable time intervals. As the reaction time is prolonged, the chemiluminescence is intensified, consistent with the formation of more hemin/G-quadruplexes. The time-dependent chemiluminescence intensities are depicted in Fig. 2B, inset. The chemiluminescence intensities level off to a saturation value after ca. four hours of telomerization. Fig. 2C depicts the chemiluminescence spectra generated by the miR-141/DSN-treated QDs that were interacted with telomerase extracted from various numbers of cells, in the presence of the dNTP mixture, for a fixed time-interval of four hours. As the telomerase content increases, the resulting chemiluminescence is intensified, consistent with the higher content of synthesized hemin/G-quadruplex catalytic units. Fig. 2C, inset, shows the resulting calibration curve, corresponding to the chemiluminescence intensities generated by telomerase extracted from different numbers of cells. The chemiluminescence intensities level off to a saturation value upon using telomerase extracted from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. The optimization of the different steps for analyzing miR-141 by the primary DSN-stimulated regeneration of the miR-141 and the secondary amplified telomerization process, enabled the sequential analysis of variable concentrations of miR-141 (Fig. 2D). As the concentration of miR-141 increased, the resulting chemiluminescence was intensified, consistent with the enhanced generation of the telomeric DNAzyme wires. Fig. 2D, inset, depicts the calibration curve corresponding to the chemiluminescence intensities generated upon analyzing various concentrations of the miR-141 by the coupled DSN/telomerase platform. The detection limit for analyzing miR-141 corresponded to 2.8 × 10−13 M. The most interesting result, however, is the impressive selectivity of the telomerase-stimulated chemiluminescence detection of miR-141 (Fig. 2E). It can be seen that the functional QDs subjected to miRs other than miR-141 do not show any chemiluminescence signal. This impressive selectivity is due to the fact that the DSN cleavage of foreign miR/(1) duplexes does not lead to the cleaved-off sequences that are recognized by telomerase. It should be noted that the analysis of miR-141 consists of a two-step procedure, where each step includes amplification mechanisms. In the first step the formation of the duplex between the probe (1) and the miR-141 provides a functional scaffold for the DSN-catalyzed regeneration of the miR-141 analyte. The cyclic cleavage of the quencher unit from the probe sites (1) provides the amplification path for the fluorescent detection of miR-141. The observed poor selectivity of this step towards miR-141 implies, however, that a fluorescence signal may only be considered as an indicator for the existence of miR-141. The second step involving the chemiluminescent, telomerase-catalyzed detection of miR-141 is very selective, and thus may be considered as a confirmatory reporting path for the quantitative detection of miR-141. We emphasize that the telomerase-stimulated chemiluminescence detection step of miR-141 includes two amplification mechanisms, i.e. the generation of long telomeric hemin/G-quadruplex catalytic chains, and the DNAzyme-catalyzed oxidation of luminol by H2O2. We further emphasize that the two-step analytical procedure is essential for the successful detection of miR-141. The first step is important to yield the primer sequence for telomerase through the amplified DSN-catalyzed regeneration of miR-141, and to provide an indicator signal for the existence of miR-141. The second step provides the amplification of the first step and the selective detection of miR-141, using chemiluminescence as a readout signal.
000 cells. The optimization of the different steps for analyzing miR-141 by the primary DSN-stimulated regeneration of the miR-141 and the secondary amplified telomerization process, enabled the sequential analysis of variable concentrations of miR-141 (Fig. 2D). As the concentration of miR-141 increased, the resulting chemiluminescence was intensified, consistent with the enhanced generation of the telomeric DNAzyme wires. Fig. 2D, inset, depicts the calibration curve corresponding to the chemiluminescence intensities generated upon analyzing various concentrations of the miR-141 by the coupled DSN/telomerase platform. The detection limit for analyzing miR-141 corresponded to 2.8 × 10−13 M. The most interesting result, however, is the impressive selectivity of the telomerase-stimulated chemiluminescence detection of miR-141 (Fig. 2E). It can be seen that the functional QDs subjected to miRs other than miR-141 do not show any chemiluminescence signal. This impressive selectivity is due to the fact that the DSN cleavage of foreign miR/(1) duplexes does not lead to the cleaved-off sequences that are recognized by telomerase. It should be noted that the analysis of miR-141 consists of a two-step procedure, where each step includes amplification mechanisms. In the first step the formation of the duplex between the probe (1) and the miR-141 provides a functional scaffold for the DSN-catalyzed regeneration of the miR-141 analyte. The cyclic cleavage of the quencher unit from the probe sites (1) provides the amplification path for the fluorescent detection of miR-141. The observed poor selectivity of this step towards miR-141 implies, however, that a fluorescence signal may only be considered as an indicator for the existence of miR-141. The second step involving the chemiluminescent, telomerase-catalyzed detection of miR-141 is very selective, and thus may be considered as a confirmatory reporting path for the quantitative detection of miR-141. We emphasize that the telomerase-stimulated chemiluminescence detection step of miR-141 includes two amplification mechanisms, i.e. the generation of long telomeric hemin/G-quadruplex catalytic chains, and the DNAzyme-catalyzed oxidation of luminol by H2O2. We further emphasize that the two-step analytical procedure is essential for the successful detection of miR-141. The first step is important to yield the primer sequence for telomerase through the amplified DSN-catalyzed regeneration of miR-141, and to provide an indicator signal for the existence of miR-141. The second step provides the amplification of the first step and the selective detection of miR-141, using chemiluminescence as a readout signal.
 | ||
| Fig. 2 Two-layer amplified detection of miR-141 by the telomerase-stimulated telomerization of a primer associated with the QDs. (A) Schematic chemiluminescence analysis of miR-141 by the two-step detection platform involving the interaction of the miR-141/(1)-modified QDs with DSN and the subsequent telomerization of domain I-modified QDs in the presence of telomerase/dNTPs. The quantitative analysis of miR-141 is then transduced by the chemiluminescence generated by the telomeric hemin/G-quadruplex DNAzyme-catalyzed oxidation of luminol by H2O2. (B) Time-dependent chemiluminescence spectra generated upon the treatment of the (1)-functionalized QDs with miR-141, 100 nM, and DSN, 0.2 U, for a fixed time-interval of one hour, and the subsequent interaction of the resulting QDs with telomerase extracted from 5000 PC-3 cells and the dNTP mixture for various time-intervals of telomerization. Inset: chemiluminescence intensities at λ = 410 nm for different time-intervals of telomerization. (C) Chemiluminescence spectra obtained upon analyzing miR-141 by applying different concentrations of telomerase in the telomerization process. The (1)-modified QDs were subjected to miR-141, 100 nM, and DSN, 0.2 U, for a fixed time-interval of one hour. The resulting QDs were then interacted with telomerase extracted from different numbers of PC-3 cells and dNTPs, and telomerization was allowed to proceed for a fixed time-interval of four hours. Inset: chemiluminescence intensities as a function of the number of PC-3 cells used for the telomerization process. At the end of the telomerization, hemin, 0.05 mM, luminol, 5 mM, and H2O2, 300 mM, were added to the system to generate chemiluminescence. The error bars indicate the standard deviation of N = 3 experiments. (D) Chemiluminescence spectra corresponding to the analysis of various concentrations of miR-141 by the (1)-functionalized CdSe/ZnS QDs, using the optimized DSN and telomerase/dNTPs conditions, as a two-layer amplification platform. The (1)-modified QDs were reacted with different concentrations of miR-141, and treated with DSN, 0.2 U, and subsequently with telomerase extracted from 10000 cells and dNTP mixture for four hours. Chemiluminescence generation conditions are detailed in the caption of Fig. 2C Inset: derived calibration curve. Error bars derived from N = 3 experiments. (E) Chemiluminescence intensity changes, ΔCL, generated upon analysis of different miRs, 100 nM each, using the (1)-functionalized QDs and the DSN/telomerase analysis scheme (conditions detailed in (D)). ΔCL = CL − CL0, where CL is the chemiluminescence intensity in the presence of the miR, and CL0 is the background luminescence in the absence of the miR. | ||
Finally, we implemented the QDs-based sensing platforms for the detection of the miR-141 biomarker in serum samples. In these experiments, miRs were extracted from the serum samples (see the Experimental section). Specifically, we applied the (1)-modified QDs for the fluorescence detection of miR-141 ( Fig. 3, panel I) and the telomerase-stimulated chemiluminescence analysis of miR-141 by the (1)-modified QDs (Fig. 3, panel II). For comparison, the same samples were subjected to analysis of the PSA, using a commercial ELISA kit. This method involves using horseradish peroxidase-labeled antibodies as catalysts for the generation of a colorimetric signal via the H2O2-stimulated oxidation of TMB to the colored benzidine, TMBox (Fig. 3, panel III). Human serum samples from PC carriers and healthy individuals were analyzed. The scatter plots corresponding to the analyses of miR-141 or PSA by the different methods are presented in Fig. 3. Using the calibration curves for the analysis of miR-141 by the CdSe/ZnS QDs (Fig. 1C, inset) and the telomerase-stimulated chemiluminescence readout (Fig. 2D), the scatter plots for the “quantitative” analysis of miR-141 in the serum samples are presented in Fig. 3, panels I and II, respectively. The results reveal several important features that should be addressed: (a) both methods reveal distinct and distinguishable concentration regions for the fluorescence or chemiluminescence intensities associated with healthy individuals or PC carriers. (b) There is an apparent discrepancy in the concentrations of miR-141 evaluated by the fluorescent QDs and by the chemiluminescence readout method. The fluorescence generated by the QDs reveals higher concentrations of miR-141 than those detected by the chemiluminescence method, in the samples from the PC carriers. While the QD method indicates miR-141 concentrations in the range of 3.8 × 10−11 M to 1.7 × 10−7 M, the same samples analyzed by the chemiluminescence method reveal a ca. 70-fold lower concentration range of miR-141, 5.3 × 10−13 M to 3.9 × 10−9 M. (c) The scatter plot corresponding to the chemiluminescence intensities generated by healthy individuals converges into a single value of (4 ± 0.2) × 10−13 M. Although the difference in the quantitative evaluation of miR-141 in the serum samples by the QDs and the chemiluminescence method is not fully understood, one may attribute the apparent higher concentrations of miR-141 determined by the QDs vs. the chemiluminescence generated by the telomeric G-quadruplex chains to several effects: (i) duplexes formed between other miRs present in the serum and the (1)-modified QDs may lead to DSN-stimulated digestive cleavage of probe (1), resulting in the cleavage of the quencher and thus increased fluorescence. The incomplete digestion of the probe does not, however, yield the primer for telomerase, and thus the foreign miRs add to the resulting fluorescence, beyond the fluorescence generated by miR-141. The effect of the foreign miRs, however, is not reflected in the chemiluminescence assay. (ii) DNases present in the serum samples induce the non-specific hydrolytic digestion of probe (1). Alternatively, DNase may partially hydrolyze the resulting telomerase primer, and thus the two processes perturb the telomerization process, resulting in a lower chemiluminescence signal. Thus, the presence of foreign miRs and DNase in the serum samples increases the fluorescence signals of the QDs beyond the “real” concentration of miR-141. In contrast, the DNase in the serum samples decreases the content of the catalytic telomeric G-quadruplexes, leading to slightly lower chemiluminescence signals as compared to analogous miR-141 concentrations in pure samples.
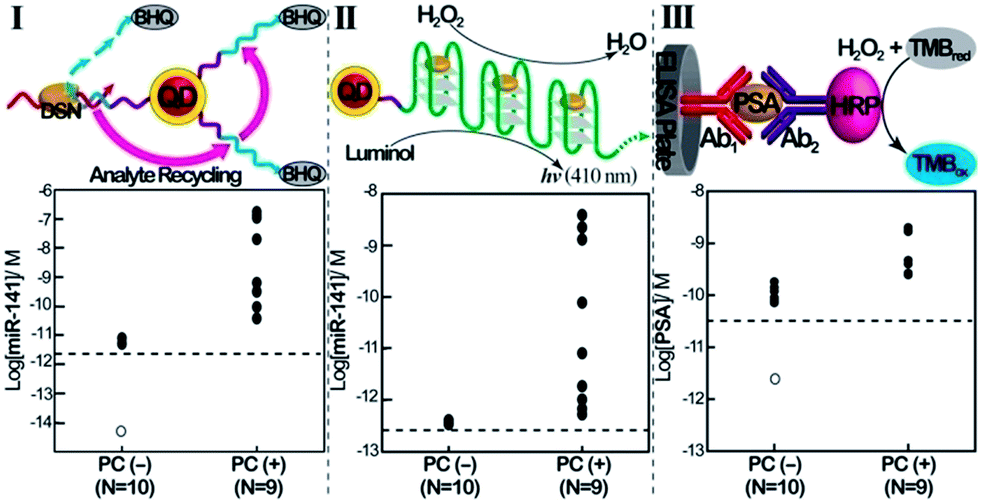 | ||
| Fig. 3 Comparison of the analysis of miR-141 and PSA in human clinical samples, using the QD/telomerase assays and PSA/ELISA immunoassay. Scatter plots corresponding to the analysis of serum samples from healthy individuals (PC (−), prostate cancer negative), N = 10, and prostate cancer carriers (PC (+), prostate cancer positive), N = 9. Panel I: concentrations of miR-141 as evaluated by the DSN-stimulated cleavage of (1)-functionalized QDs. Panel II: concentrations of miR-141 in the samples as evaluated using chemiluminescence generated by the two-step DSN/telomerase-dNTPs analysis platform. Panel III: concentrations of PSA as evaluated by the standard immunoassay. Horizontal dotted lines represents the corresponding detection limits for each assay. Open circles represent values <LOD (N = 7 in panel I; N = 3 in panel III). The tabulation of data is shown in ESI Table S1.† | ||
Conclusions
The present study has introduced a two-step analytical platform for the detection of the miR-141 prostate cancer biomarker. The first step involves the use of CdSe/ZeS QDs modified with quencher-functionalized nucleic acid probes, as an optical label for the fluorescent detection of miR-141. The hybridization of miR-141 with the probe units activates the DSN-stimulated cleavage of the resulting duplex, the removal of the quencher units, and the regeneration of miR-141 to form an amplification path. Due to the limited selectivity of the first step of the analytical platform, the resulting fluorescence of the QDs could serve only as an “indicator” signal for the existence of miR-141. The non-selective first-step of the analytical platform is, however, essential for the overall sensing process, since it generates, by a DSN-stimulated cleavage process, the primer units for the selective second-step, an analysis scheme involving telomerase as a means of amplification. It should be noted that the use of nanoparticles (e.g. CdSe/ZnS QDs) in the first-step detection scheme is important, since the nanoparticles act as carriers for many probe units that allow the effective utilization of the DSN-regenerated miR-141, and the carriage of many primer units for the second-step telomerase-mediated reaction. The use of CdSe/ZnS QDs as nanocarriers for the sensing process is particularly interesting since the nanoparticles are conjugated to hemin/G-quadruplex, chemiluminescence generating, telomeric DNAzyme chains. This would allow, in the future, the use of a chemiluminescence resonance energy transfer (CRET) process as a readout signal.25 Particularly important is the successful implementation of the sensing platform to analyze real serum samples. The validation of the present method by comparing it to reference gene analysis technologies31 is still a future challenge.Experimental
Chemicals and reagents
NaCl (sodium chloride), MgCl2 (magnesium chloride), KCl (potassium chloride), NaOH (sodium hydroxide), CHCl3 (chloroform), K2HPO4 (potassium phosphate dibasic), KH2PO4 (potassium phosphate monobasic), 35% HCl (hydrochloric acid), 35% H2O2 (hydrogen peroxide), HEPES (4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid), DMSO (dimethyl sulfoxide), PMSF (phenylmethanesulfonyl fluoride), DTT (DL-dithiothreitol), GSH (L-glutathione reduced), MeOH (methanol), Tris–HCl (trizma hydrochloride), hemin, spermidine, glycerol, and luminol were purchased from Sigma-Aldrich (St. Louis, MO, USA). Triton-X100 (iso-octylphenoxypolyethoxyethanol) was obtained from Thermo Fisher Scientific (MA, USA). EDTA (ethylene diaminetetra acetate) was acquired from Amersham Pharmacia Biotech (St. Albans, UK). BS3 (bis[sulfosuccinimidyl]suberate) was obtained from Pierce Biotechnology (Rockford, IL, USA). CdSe/ZnS quantum dots (QD) (10 mg mL−1) in toluene were purchased from NN-LABS (LLC., Fayetteville, AR). Duplex-specific nuclease (DSN) was acquired from Evrogen Joint Stock company (Moscow, Russia). RNase inhibitor and dNTP were purchased from Roche (Indianapolis, Indiana, USA). Human miRNeasy serum/plasma kit (Cat. no. 217184) was obtained from Qiagen (Valencia, CA). Human prostate-specific antigen ELISA kit (Cat. no. 55R-BC2601) was obtained from Fitzgerald Industries International (MA, USA). All aqueous solutions were prepared using distilled and deionized water (d.d. water, resistivity > 18 MΩ cm) purified with a Milli-Q water purification system (Bedford, MA). All pipette tips and tubes were RNase-free and used without further RNase inactivation. All glassware was oven baked at 250 °C for 4 h before use to inactivate any RNase. Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). All oligonucleotides were used as provided and diluted with 10 mM of phosphate buffer solution (pH 7) to obtain a stock solution of 100 μM.The sequences of the oligomers used in this study are as follows:
(1) Probe DNA: 5′-NH2(CH2)6TTA GGG CCA TCT TTA CCA GAC AGT GTT A-BHQ2-3′.
(2) miR-141: 5′-UAACACUGUCUGGUAAAGAUGG-3′.
(3) miR-200b: 5′-UAAUACUGCCUGGUAAUGAUGA-3′.
(4) miR-200a: 5′-UAACACUGUCUGGUAACGAUGU-3′.
(5) miR-21: 5′-UAGCUUAUCAGACUGAUGUUGA-3′.
(6) let-7a: 5′-UGAGGUAGUAGGUUGUAUAGUU-3′.
Preparation of GSH-functionalized CdSe/ZnS quantum dots
The GSH-functionalized quantum dots (GSH-QDs) were prepared based on the modified ligand exchange procedure.25 Functionalization was initiated by transferring the QDs from toluene to CHCl3; 20 μL of CdSe/ZnS core/shell octadecylamine (ODA)-capped QDs were precipitated from the stock solution upon addition of 0.8 mL of MeOH. This was followed by centrifugation at 3000 rpm for 5 min, and the toluene supernatant was then decanted and the resulting precipitate re-suspended in 0.4 mL of CHCl3. To substitute ODA ligand for GSH ligand, 1 mL of GSH alkaline methanol solution (40 mg mL−1 in MeOH containing KOH, 20 mg mL−1) was slowly added to the QD solution (in CHCl3) at an ambient temperature. The resulting mixture was reacted at 50 °C for 2 h and subsequently cooled down to 27 °C, and was then allowed to react overnight. After the addition of 1 mL of 1 mM NaOH aqueous solution, GSH-decorated QDs were partitioned to the upper aqueous phase, indicating a successful exchange of the ligands. Finally, methanol was added to the aqueous phase containing GSH-functionalized QDs, followed by centrifugation at 3000 rpm for 3 min to remove unreacted GSH. The purified GSH-functionalized QDs were subsequently re-suspended in 200 μL of d.d. water and stored at 4 °C prior to use.Modification of GSH-functionalized QDs with nucleic acid
The GSH-functionalized QDs were first reacted with BS3 in HEPES buffer (100 mM, pH 7) for 20 min at an ambient temperature,32 followed by precipitation with the addition of 200 μL of MeOH and 3 mg of NaCl to remove the excess of free BS3. To the resulting purified QDs, 50 μL of the probe (1) (10 mM, in phosphate buffer, pH 7) was added, and the reaction mixture was shaken for 2 h. The purification steps using MeOH and NaCl as described above were repeated twice. The probe (1)-modified QDs were re-suspended in 200 μL of HEPES buffer (100 mM, pH 7) and stored at 4 °C before use.Determination of the loading of the GSH-functionalized QDs with the probe DNA (1)
The absorption spectrum of a known concentration of non-modified QDs (GSH-functionalized QDs) was recorded prior to the modification of the QDs with the probe DNA. Normalization was then carried out to the same OD value at λ = 620 nm recorded for the non-modified QDs, since DNA does not absorb light at 620 nm. The subtraction of the spectrum of the modified QDs from that of the non-modified QDs yields the absorbance difference at λ = 620 nm, which allows the calculation of the probe DNA concentration. Knowing the concentration of QDs and the concentration of the probe DNA, the loading of the probe DNA on the QDs was evaluated using a Cary 300 Bio UV-Vis spectrophotometer (Varian, Mulgrave Victoria, Australia).Preparation of telomerase extract
PC-3 cells cultured in Ham's F12K medium with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin were obtained from the Culture Collection and Research Center (CCRC) (Hsinchu, Taiwan). The cells were grown with fresh medium on a 75 cm2 flask at 37 °C in a humidified 5% CO2 incubator. After being harvested with trypsin, cells were centrifuged at 5000 rpm for 5 min to remove the medium. The telomerase in the resulting PC-3 cell pellet was extracted according to the protocol reported by Cohen and Reddel.33Isolation of microRNA from clinical samples
All serum samples from prostate cancer carriers and healthy individuals were collected by the team of Dr Yen-Chuan Ou at the Division of Urology, Taichung Veterans General Hospital. All samples analyzed in the current study were appropriately consented to and collected using IRB-approved clinical protocols from donors with prostate cancer, and matched. Circulating miR-141 in serum was extracted using the human miRNeasy serum/plasma kit according to the manufacturer's instruction. Briefly, 1 mL of Qiazol lysis reagent and 20 U of RNase inhibitor were added to 0.2 mL of serum and mixed by pipetting, followed by incubation at an ambient temperature for 5 min to dissociate the nucleoprotein complexes. After adding 0.2 mL of CHCl3, the mixture was vigorously vortexed for 15 s and incubated at room temperature for 3 min, followed by centrifugation at 12000g at 4 °C for 15 min. The upper aqueous phase containing microRNA was obtained, to which 1.5 volumes of 95% EtOH were added and mixed by pipetting again. Finally, purification of the microRNA was carried out using a spin-column format, as described in the protocol. The purified microRNA extracted from each clinical serum sample was eluted with double-distilled water (14 μL) and stored at −20 °C before analysis.
Analysis of microRNA by the fluorescence method using the first step involving the QDs
The fluorescence approach for the analysis of miR-141 was performed by preparing a mixture consisting of 30 μL of DSN (0.2 U), (1)-modified QDs (33.3 nmol) and the target microRNA (5 μL of the specified concentration) in 50 mM of a Tris–HCl buffer solution containing 5 mM MgCl2 and 1 mM DTT, pH 8. This was followed by incubation at 55 °C for 1 h. The reaction volume was brought up to 100 μL with the same buffer solution and the fluorescence changes were recorded using a Varian Cary Eclipse fluorescence spectrometer (Varian, Mulgrave Victoria, Australia).Analysis of microRNAs by the two-step analysis process involving the QD/telomerase and chemiluminescence as a readout signal
The chemiluminescence approach for the analysis of miR-141 was performed by preparing 30 μL of a solution mixture consisting of DSN (0.2 U), (1)-modified QDs (33.3 nmol) and the target microRNA (5 μL of the specified concentration) in 50 mM of Tris–HCl buffer solution containing 5 mM MgCl2 and 1 mM DTT, pH 8, and this was incubated at 55 °C for 1 h. The telomerization reaction was initiated by adding 20 μL of a dNTP mixture containing 0.2 mM each of dNTP, telomerase (extracted from 104 PC-3 cells), 300 mM KCl, and 1 mM spermidine to the QDs mixture, and the system was allowed to react at 37 °C for 4 h. After telomerization, the resulting mixture was treated with 1 μL of 0.05 mM hemin for another 30 min at an ambient temperature. Subsequently 25 μL of luminol (5 mM) and 25 μL of H2O2 (300 mM) were quickly added, and the chemiluminescence emission intensity was immediately measured using a photon-counting spectrometer (Edinburgh Instruments, F900).Analysis of human prostate-specific antigen (PSA) in clinical serum samples
The analysis of human PSA in clinical serum samples was performed using a commercial ELISA kit according to the manufacturer's instruction. The absorbance changes at 450 nm were measured by a 96-well microplate reader (Sunrise, Tecan Trading AG, Switzerland).Acknowledgements
I. Willner is supported by the Israel Science Foundation and J. A. Ho is supported by the Taiwan Ministry of Science and Technology, under contracts 99-2923-M-002-008-MY2, 101-2113-M-002-003-MY3, and 102-2628-M-002-004-MY4. We thank Dr F. Wang and Mrs E. Sharon for their help in formulating the paper.Notes and references
- A. Jemal, R. Siegel, J. Xu and E. Ward, Ca-Cancer J. Clin., 2010, 60, 277 CrossRef PubMed.
- F. Schröder and M. Roobol-Bouts, Curr. Opin. Urol., 2009, 19, 227 CrossRef PubMed.
- G. L. Kirsten, M. V. Maxwell, E. P. Eric, C. R. Matthew, P. J. David, K. W. Michael, W. Katrine and C. R. Peter, J. Urol., 2004, 171, 2255 CrossRef.
- J. W. F. Catto, S. Miah, H. C. Owen, H. Bryant, K. Myers, E. Dudziec, S. Larré, M. Milo, I. Rehman, D. J. Rosario, E. Di Martino, M. A. Knowles, M. Meuth, A. L. Harris and F. C. Hamdy, Cancer Res., 2009, 69, 8472 CrossRef CAS PubMed.
- C. Blenkiron, L. D. Goldstein, N. P. Thorne, I. Spiteri, S.-F. Chin, M. J. Dunning, N. L. Barbosa-Morais, A. E. Teschendorff, A. R. Green, I. O. Ellis, S. Tavaré, C. Caldas and E. A. Miska, GenomeBiology, 2007, 8, R214 CrossRef PubMed.
- P. S. Mitchell, R. K. Parkin, E. M. Kroh, B. R. Fritz, S. K. Wyman, E. L. Pogosova-Agadjanyan, A. Peterson, J. Noteboom, K. C. O'Briant, A. Allen, D. W. Lin, N. Urban, C. W. Drescher, B. S. Knudsen, D. L. Stirewalt, R. Gentleman, R. L. Vessella, P. S. Nelson, D. B. Martin and M. Tewari, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10513 CrossRef CAS PubMed.
- S. Liu, Y. Wang, J. Ming, Y. Lin, C. Cheng and F. Li, Biosens. Bioelectron., 2013, 49, 472 CrossRef CAS PubMed.
- S. Shimron, F. Wang, R. Orbach and I. Willner, Anal. Chem., 2012, 84, 1042 CrossRef CAS PubMed.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149 CrossRef CAS PubMed.
- Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowarski, M. Kotler and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 7384 CrossRef CAS PubMed.
- R. Orbach, L. Mostinski, F. Wang and I. Willner, Chem.–Eur. J., 2012, 18, 14689 CrossRef CAS PubMed.
- F. Wang, L. Freage, R. Orbach and I. Willner, Anal. Chem., 2013, 85, 8196 CrossRef CAS PubMed.
- Y. Tian, Y. He and C. Mao, ChemBioChem, 2006, 7, 1862 CrossRef CAS PubMed.
- Y. Wen, Y. Xu, X. Mao, Y. Wei, H. Song, N. Chen, Q. Huang, C. Fan and D. Li, Anal. Chem., 2012, 84, 7664 CrossRef CAS PubMed.
- B. Li, Y. Jiang, X. Chen and A. D. Ellington, J. Am. Chem. Soc., 2012, 134, 13918 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275 CrossRef CAS PubMed.
- Z. Lin, W. Yang, G. Zhang, Q. Liu, B. Qiu, Z. Cai and G. Chen, Chem. Commun., 2011, 47, 9069 RSC.
- T. Tian, H. Xiao, Z. Zhang, Y. Long, S. Peng, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem.–Eur. J., 2013, 19, 92 CrossRef CAS PubMed.
- S. Bi, J. Zhang and S. Zhang, Chem. Commun., 2010, 46, 5509 RSC.
- X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816 CrossRef CAS PubMed.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602 CrossRef CAS PubMed.
- M. Frasco and N. Chaniotakis, Sensors, 2009, 9, 7266 CrossRef CAS PubMed.
- H. Peng, L. Zhang, T. H. M. Kjällman and C. Soeller, J. Am. Chem. Soc., 2007, 129, 3048 CrossRef CAS PubMed.
- A. Niazov, R. Freeman, J. Girsh and I. Willner, Sensors, 2011, 11, 10388 CrossRef CAS PubMed.
- R. Freeman, X. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133, 11597 CrossRef CAS PubMed.
- E. Sharon, R. Freeman and I. Willner, Anal. Chem., 2010, 82, 7073 CrossRef CAS PubMed.
- R. Freeman, X. Liu and I. Willner, Nano Lett., 2011, 11, 4456 CrossRef CAS PubMed.
- S. B. Cohen, M. E. Graham, G. O. Lovrecz, N. Bache, P. J. Robinson and R. R. Reddel, Science, 2007, 315, 1850 CrossRef CAS PubMed.
- T. M. Bryan and T. R. Cech, Curr. Opin. Cell Biol., 1999, 11, 318 CrossRef CAS.
- R. Freeman, E. Sharon, C. Teller, A. Henning, Y. Tzfati and I. Willner, ChemBioChem, 2010, 11, 2362 CrossRef CAS PubMed.
- G. K. Geiss, et al. , Nat. Biotechnol., 2008, 26, 317 CrossRef CAS PubMed.
- R. Freeman, J. Girsh, A. F. Jou, J. A. Ho, T. Hug, J. Dernedde and I. Willner, Anal. Chem., 2012, 84, 6192 CrossRef CAS PubMed.
- S. B. Cohen and R. R. Reddel, Nat. Methods, 2008, 5, 355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of detection conditions and tabulation of data in Fig. 3. See DOI: 10.1039/c4sc02104e1 |
| This journal is © The Royal Society of Chemistry 2015 |