
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chelation-induced diradical formation as an approach to modulation of the amyloid-β aggregation pathway†
Meghan R.
Porter
a,
Akiko
Kochi
bc,
Jonathan A.
Karty
a,
Mi Hee
Lim
*cd and
Jeffrey M.
Zaleski
*a
aDepartment of Chemistry, Indiana University, Bloomington, Indiana 47405, USA. E-mail: zaleski@indiana.edu
bDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, USA
cDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 689-798, Korea. E-mail: mhlim@unist.ac.kr
dLife Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, USA
First published on 30th October 2014
Abstract
Current approaches toward modulation of metal-induced Aβ aggregation pathways involve the development of small molecules that bind metal ions, such as Cu(II) and Zn(II), and interact with Aβ. For this effort, we present the enediyne-containing ligand (Z)-N,N′-bis[1-pyridin-2-yl-meth(E)-ylidene]oct-4-ene-2,6-diyne-1,8-diamine (PyED), which upon chelation of Cu(II) and Zn(II) undergoes Bergman-cyclization to yield diradical formation. The ability of this chelation-triggered diradical to modulate Aβ aggregation is evaluated relative to the non-radical generating control pyridine-2-ylmethyl-(2-{[(pyridine-2-ylmethylene)-amino]-methyl}-benzyl)-amine (PyBD). Variable-pH, ligand UV-vis titrations reveal pKa = 3.81(2) for PyBD, indicating it exists mainly in the neutral form at experimental pH. Lipinski's rule parameters and evaluation of blood–brain barrier (BBB) penetration potential by the PAMPA–BBB assay suggest that PyED may be CNS+ and penetrate the BBB. Both PyED and PyBD bind Zn(II) and Cu(II) as illustrated by bathochromic shifts of their UV-vis features. Speciation diagrams indicate that Cu(II)–PyBD is the major species at pH 6.6 with a nanomolar Kd, suggesting the ligand may be capable of interacting with Cu(II)–Aβ species. In the presence of Aβ40/42 under hyperthermic conditions (43 °C), the radical-generating PyED demonstrates markedly enhanced activity (2–24 h) toward the modulation of Aβ species as determined by gel electrophoresis. Correspondingly, transmission electron microscopy images of these samples show distinct morphological changes to the fibril structure that are most prominent for Cu(II)–Aβ cases. The loss of CO2 from the metal binding region of Aβ in MALDI-TOF mass spectra further suggests that metal–ligand–Aβ interaction with subsequent radical formation may play a role in the aggregation pathway modulation.
Introduction
Alzheimer's disease (AD) is the most common form of dementia, affecting over 24 million people worldwide.1 It is estimated that this number will nearly double by 2030, partly due to demographic aging resulting from improved healthcare.1 The disease presence and progression are pathologically characterized by accumulation of misfolded amyloid-β (Aβ) peptides deriving from β- and γ-secretase cleavage of the amyloid precursor protein (APP)2–4 to produce Aβ40 and Aβ42 that self-assemble through hydrophobic interactions to form oligomers, protofibrils, fibrils, and ultimately, insoluble plaques.4–6 It has been proposed that Aβ plaque accumulation may arise from an imbalance in Aβ production and clearance (i.e., amyloid cascade hypothesis);4,7–9 accumulation of these peptides alone can impair neuronal mitochondrial function, leading to oxidative stress, inflammation, and the neurodegeneration commonly associated with AD (i.e., oxidative stress hypothesis).4,10–12 In addition to self-aggregation, miscompartmentalization and dyshomeostasis of metals are found in AD-afflicted brains. In particular, elevated levels of metals, such as Cu, Zn, and Fe, are observed in Aβ plaques.3–5,13–19 Metal binding to Aβ is shown to facilitate peptide aggregation and in the case of redox active metal ions, reactive oxygen species (ROS) can be generated via Fenton-like reactions, leading to oxidative stress.4,5,17,19–24On the basis of the observed metal ion dyshomeostasis, metal–Aβ interaction, and metal-involved Aβ reactivity, there has been considerable interest in the development of metal chelators capable of regulating metal ion distribution distribution and amyloid pathology. For example, the hydroxyquinoline-based antifungal drug clioquinol (CQ) decreased Aβ deposits and showed improved cognition in Phase II clinical trials for AD, in part due to its ability to inhibit binding of Zn(II) and Cu(II) to Aβ via chelation.25–27 Moreover, the second generation 8-hydroxyquinoline ionophore PBT2 also improved learning and memory by redistributing Cu(II) and Zn(II) and lowered cerebrospinal fluid levels of Aβ in Phase II clinical trials.28,29
Although CQ and PBT2 have presented noticeable effects on metal redistribution and Aβ clearance, the relationship between metal-associated Aβ (metal–Aβ) species and AD pathogenesis is still unclear, thus new efforts on developing chemical tools for specifically studying metal–Aβ species have been made.21,24,30–33 For example, the rational design of chelators containing dimethylaniline and polydentate motifs using nitrogen and oxygen donor atoms for metal ions has led to blood–brain barrier (BBB) permeable compounds that modulate metal-induced Aβ aggregation, reduce Cu–Aβ ROS formation, demonstrate antioxidant activity, and/or decrease metal–Aβ toxicity in vitro.30,32–34
Our latest approach to bifunctional chelators for Aβ modification derives from drugs such Fe-Bleomycin or hydroxyl radical footprinting reagents that act via Fenton chemistry and perform H-atom abstraction from the ribose ring of DNA leading to strand scission.35,36 Similarly, enediyne natural products such as calicheamicin that generate a potent 1,4-phenyl diradical also affect strand scission by H-atom abstraction. Radical reactions of these types however, are not limited to DNA substrates. Rather, radical-mediated footprinting is an established methodology for evaluating protein structure via solvent accessible reactivity,37 as well as for mapping protein–protein and protein–DNA interactions.38–47 Generation of ROS by reaction with redox active Fe, Cu, and Mn-complexes38,48–58 in the presence of reductant leads to controlled backbone or side chain attack which can be used to evaluate regions of macromolecular interface. While ribose ring radical strand breaks in DNA are generally due to H-atom abstraction from relatively weak tertiary C–H bonds38 that are statistically plentiful and readily accessible,59 H-atom abstraction from proteins is more complex. Direct H-abstraction from the α-carbon and side chain-assisted H-abstraction both lead to backbone cleavage,59–61 but poor solvent permeability and the statistical probability of extensive side chain oxidation make this process less prevalent.37,39,46,47,59,62 Somewhere between these limits lies calicheamicin which performs α-H abstraction from the protector protein CalC at Gly-113, cleaving the protein in a radical self-resistance mechanism.63
With this backdrop, we envisioned a bifunctional agent that could attack Aβ aggregates by initially chelating Aβ-bound metal ions to disrupt the peptide structure and subsequently using this chelation event to induce diradical formation that would further modify the remaining Aβ aggregates. We have shown that the compound (Z)-N,N′-bis[1-pyridin-2-yl-meth(E)-ylidene]oct-4-ene-2,6-diyne-1,8-diamine (PyED) (Fig. 1) binds a wide array of metal ions such as Mg,64 Cu, Fe, and Zn and these complexes may be thermally activated to yield a potent 1,4-diradical intermediate. Our experience with enediyne activation via metal coordination64–66 and photochemical67–69 diradical formation has taught us that these molecular frameworks are capable of both H-atom abstraction64,66,67 and addition/polymerization reactions68,69 depending upon the substrate and radical–radical coupling proximity. Additionally, PyED has demonstrated enhanced activity under clinically relevant hyperthermic conditions (42.5 °C).70 Although hyperthermic treatments have not commonly been applied in the field of AD, hyperthermia has been established as a method to enhance therapeutic efficiency when used in combination with other cancer treatments both in vitro71–75 and in vivo75–78 (≤45.5 °C). Thus, herein we report the application of such reactions to metal-bound (Cu(II), Zn(II)) Aβ aggregates by administration of PyED at physiological (37 °C) and hyperthermic (43 °C) temperatures relative to the non-radical generating control pyridine-2-ylmethyl-(2-{[(pyridine-2-ylmethylene)-amino]-methyl}-benzyl)-amine, PyBD (Fig. 1).
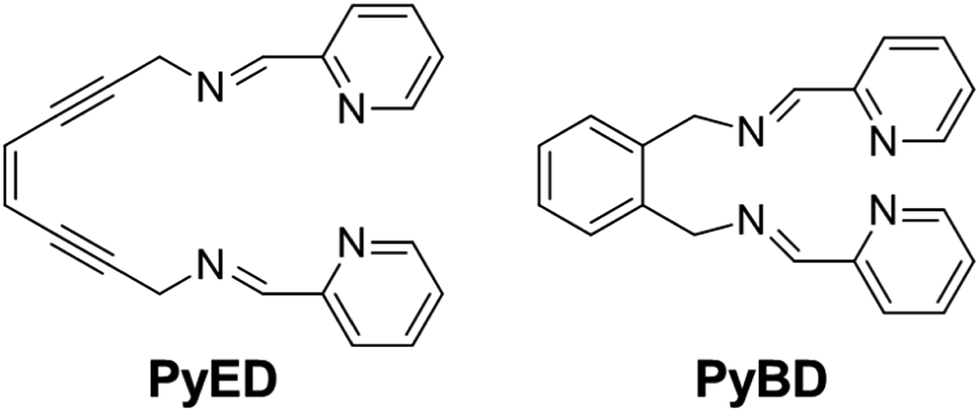 | ||
| Fig. 1 Structures of radical-generating enediyne and cyclized control ligands employed for modulation of Aβ species. | ||
Results and discussion
Rationale and characterization of PyED and PyBD used for modulating metal–Aβ species
The reactive compounds PyED (chelation + radical generation) and PyBD (chelation alone) for the modulation of Aβ species were synthesized and characterized by their 1H, 13C NMR, and mass signatures according to literature precedent.64 Cyclization of PyED by Cu(II) or Zn(II) chelation was investigated in MeOH and occurs within 4 h at 37 °C upon radical trapping with 1,4-cyclohexadiene and extraction with NaBH4 (12 equiv.)/EDTA (pH 10.6).64 The 13C NMR feature at δ 128 ppm and ESI-MS (m/z: 319.2) are diagnostic of cyclized product formation indicating PyED undergoes rapid radical formation in the presence of Cu(II) or Zn(II). Variable-pH UV-visible (UV-vis) titrations were conducted to evaluate the protonation state of the ligand in solution, particularly at physiologically relevant pH (pH = 7.4).34,79,80 In light of the fact that PyED slowly generates reactive radicals at ambient temperature over the timescale of the measurement (4–5 h), speciation was determined using the nonreactive, cyclized control PyBD. Titration results indicate a single acid ionization constant (pKa) for PyBD (pKa = 3.81(2)), suggesting that neutral and monoprotonated forms of the ligand may be present in solution depending on pH. Furthermore, the solution speciation diagram reveals that PyBD is expected to exist mainly in the neutral form at pH 7.4 (Fig. 2).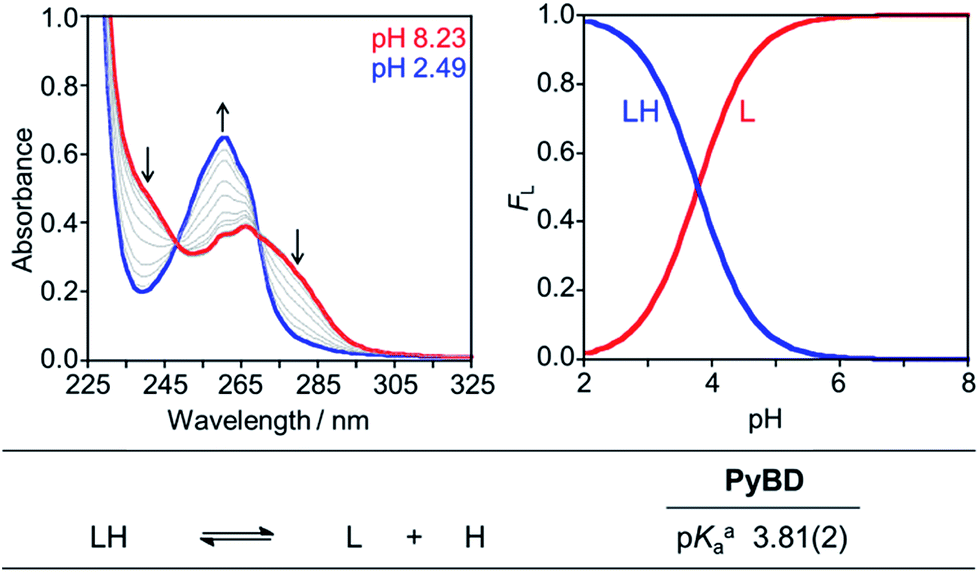 | ||
| Fig. 2 Solution speciation of PyBD (50 μM). Left: UV-vis spectra in the range of pH 2–8. Right: solution speciation diagram (FL = fraction of compound in given protonation state). Bottom: acidity constants of L (L = PyBD) with charges omitted for clarity. Speciation was performed at room temperature with I = 0.1 M NaCl. aError in the last digit is indicated in parentheses. | ||
In an effort to establish the drug-likeness of PyED and its potential to penetrate the BBB, Lipinski's rule parameters (MW < 450, c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP < 5.0, HBA < 10, HBD < 5) and the logBB were evaluated (Table 1).81–84 The resulting values (MW = 312.37, clogP = 1.01, HBA = 4, HBD = 0) indicate PyED has drug-like characteristics as well as possible BBB permeability (Table 1). In order to verify the predicted ability of PyED to penetrate the BBB, an in vitro PAMPA–BBB assay was performed following literature procedure.34,81,85 Using the empirical classification for BBB-permeable molecules, the measured permeability value, −logPe, for PyED (−logPe = 4.9 ± 0.1) suggests PyED may be likely to penetrate the BBB.
logP < 5.0, HBA < 10, HBD < 5) and the logBB were evaluated (Table 1).81–84 The resulting values (MW = 312.37, clogP = 1.01, HBA = 4, HBD = 0) indicate PyED has drug-like characteristics as well as possible BBB permeability (Table 1). In order to verify the predicted ability of PyED to penetrate the BBB, an in vitro PAMPA–BBB assay was performed following literature procedure.34,81,85 Using the empirical classification for BBB-permeable molecules, the measured permeability value, −logPe, for PyED (−logPe = 4.9 ± 0.1) suggests PyED may be likely to penetrate the BBB.
| Calculationa | PyED | Lipinski rule parameters and others |
|---|---|---|
|
a MW, molecular weight; clogP, calculated logarithm of the octanol–water partition coefficient; HBA, hydrogen-bond acceptor atoms; HBD, hydrogen-bond donor atoms; PSA, polar surface area; logBB = −0.0148 × PSA + 0.152 × clogP 0.130.
b The values of −logPe were measured by the parallel artificial membrane permeability assay (PAMPA).
c CNS+ compounds have the ability to permeate the BBB and target the CNS, while CNS− compounds have poor permeability through the BBB and therefore, their bioavailability into the CNS is considered minimal.
|
||
| MW | 312.37 | 450 |
|
clogP |
1.01 | 5.0 |
| HBA | 4 | 10 |
| HBD | 0 | 5 |
| PSA | 50.5 | 90 Å2 |
| logBB |
−0.464 | 0.3 (readily crosses the BBB) |
| −1.0 (poorly distributed in the brain) | ||
| −logPeb |
4.9 ± 0.1 | |
| CNS± predictionc | CNS+ | −logPe 5.4 (CNS+) |
| −logPe 5.7 (CNS−) |
||
Metal binding properties of PyED and PyBD
Divalent metal binding of PyED and PyBD at 0 °C was demonstrated by bathochromic shifts of their UV-vis features (PyEDλ = 264 nm; PyBDλ = 272 nm) upon addition of ZnCl2 or CuCl2 (1 equiv.) in ethanol (Fig. S1†). At 1 equiv. of MCl2, the absorption spectra of PyBD show the formation of distinct metallated species with larger bathochromic shifts observed for Cu(II) binding relative to Zn(II). For the more flexible chelate PyED, these shifts are somewhat less pronounced and indicate parallel, but slightly weaker ligand binding under these unactivating conditions (0 °C). The apparent trend of enhanced Cu(II) binding relative to Zn(II) is consistent with those observed for N-donor functionalities within a range of flexible ligands.25,86–88Although the neutral form of PyBD is the major species in solution at physiological pH (vide supra), variable-pH UV-vis titrations were also conducted to elucidate complexation and binding properties of PyBD with Cu(II) in solution at ambient temperature and the proposed local pH for Cu(II)–Aβ species (pH = 6.6) (Fig. 3, left). Based on the pKa value determined for PyBD and these titration results, the stability constants (logβ) for the these complexes were determined to be 12.2(8) and 4.4(8) for CuL and Cu(LH), respectively. A solution speciation diagram was modeled using these stability constants and suggests complexation of PyBD with Cu(II) occurs in a 1:1 metal:ligand ratio. While neutral and protonated forms of Cu(II)–PyBD may exist at different pH values, the data indicate that the neutral Cu(II)–PyBD form is the major species at pH 6.6 (Fig. 3, right). Additionally, the concentration of free Cu(II) in solution at pH 6.6 yields a pCu value of 8.3(4) (pCu = −log[Cu(II)]unbound). The pCu magnitude suggests an approximate Kd for Cu(II)–PyBD to be ca. nanomolar. When considered with the reported Kd values for Cu(II)–Aβ species (picomolar to nanomolar range),4,5,17,19–21,24,89 this approximate dissociation constant indicates that PyBD may be able to compete for Cu(II) binding in Cu(II)–Aβ species.
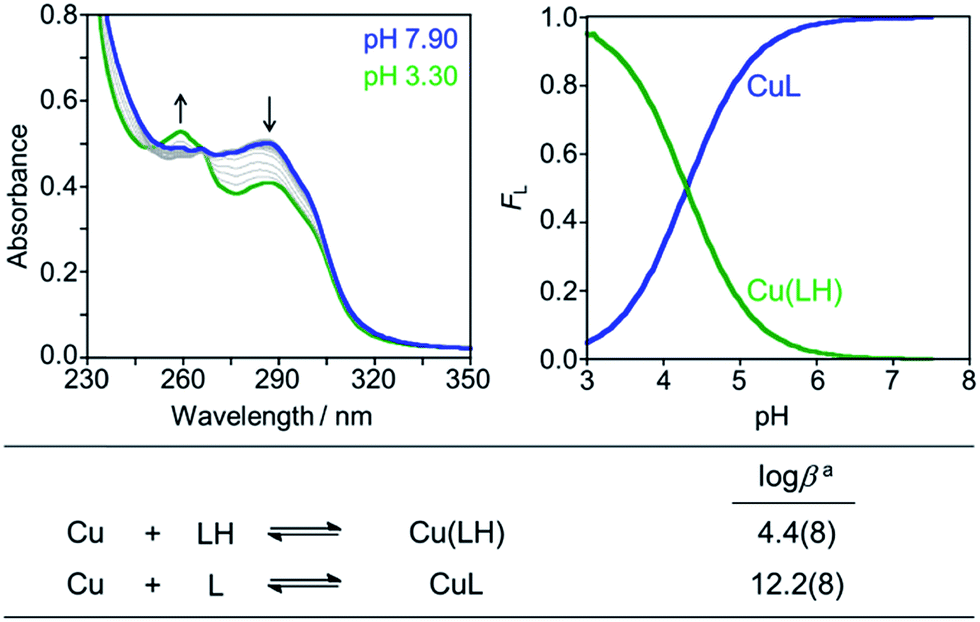 | ||
| Fig. 3 Solution speciation of the Cu(II)–PyBD complex. Left: UV-vis spectra in the range of pH 3–8 ([Cu(II)]/[PyBD] = 1:1; [Cu(II)]total = 50 μM). Right: solution speciation diagram (FCu = fraction of free Cu and Cu complexes). Bottom: stability constants of the Cu(II)–PyBD complex with charges admitted for clarity. Titrations were performed at room temperature with I = 0.1 M NaCl. aError in the last digit is indicated in parentheses. | ||
To estimate the ability of PyED to bind Cu(II) in the presence of other biologically relevant metal ions such as Ca(II), Co(II), Fe(II), Fe(III), Mg(II), Mn(II), Ni(II), and Zn(II), selectivity was evaluated for the unreactive model compound PyBD by a competitive UV-vis absorption assay (Fig. S2†). Even in the presence of a large excess of competing metal ion, PyBD displays good selectivity for Cu(II) over Ca(II), Co(II), Mg(II), Mn(II), and Ni(II), while significant binding is shown in the presence of Fe(II) and Fe(III) (Fig. S2†). The observation that PyBD demonstrates selectivity for Cu(II) over Zn(II) leads to the expectation that modulation of Cu(II)-bound Aβ species via metal chelation and subsequent radical generation by PyED may be more prominent than for the Zn(II)-bound species (vida infra). Overall, the tetradentate pyridine–imine binding moiety of PyBD and PyED may be desirable for reacting with Cu(II)–Aβ species over other biologically relevant divalent metal ions.
Effect of PyED and PyBD on metal-free and metal-triggered Aβ aggregation in vitro
In order to assess the ability of bifunctional PyED to modulate metal-induced Aβ40 and Aβ42 aggregation pathways, in vitro disaggregation and inhibition experiments were conducted (Scheme 1).32,79,80 For comparison to PyED reactivity, the influence of monofunctional PyBD on Aβ aggregation was also examined. Disaggregation assays were designed to investigate the potential of both PyED and PyBD to structurally alter preformed metal-free and metal-associated Aβ aggregates (Fig. 4 and 5), while inhibition experiments probed the compounds' ability to control the formation of metal-free and metal-induced Aβ aggregates (Fig. S3 and S4†). The resultant Aβ species were characterized using gel electrophoresis followed by Western blot with an anti-Aβ antibody (6E10), and morphological changes were monitored by transmission electron microscopy (TEM).32,80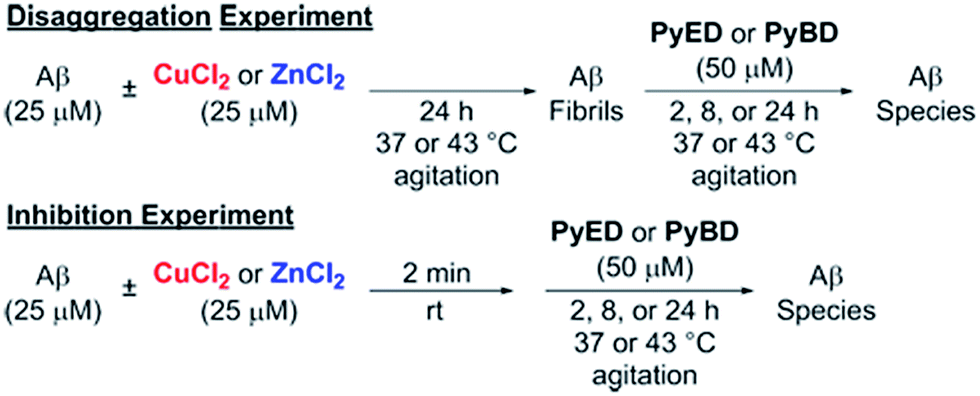 | ||
| Scheme 1 Experimental set-up for Aβ40 and Aβ42 disaggregation and inhibition assays. | ||
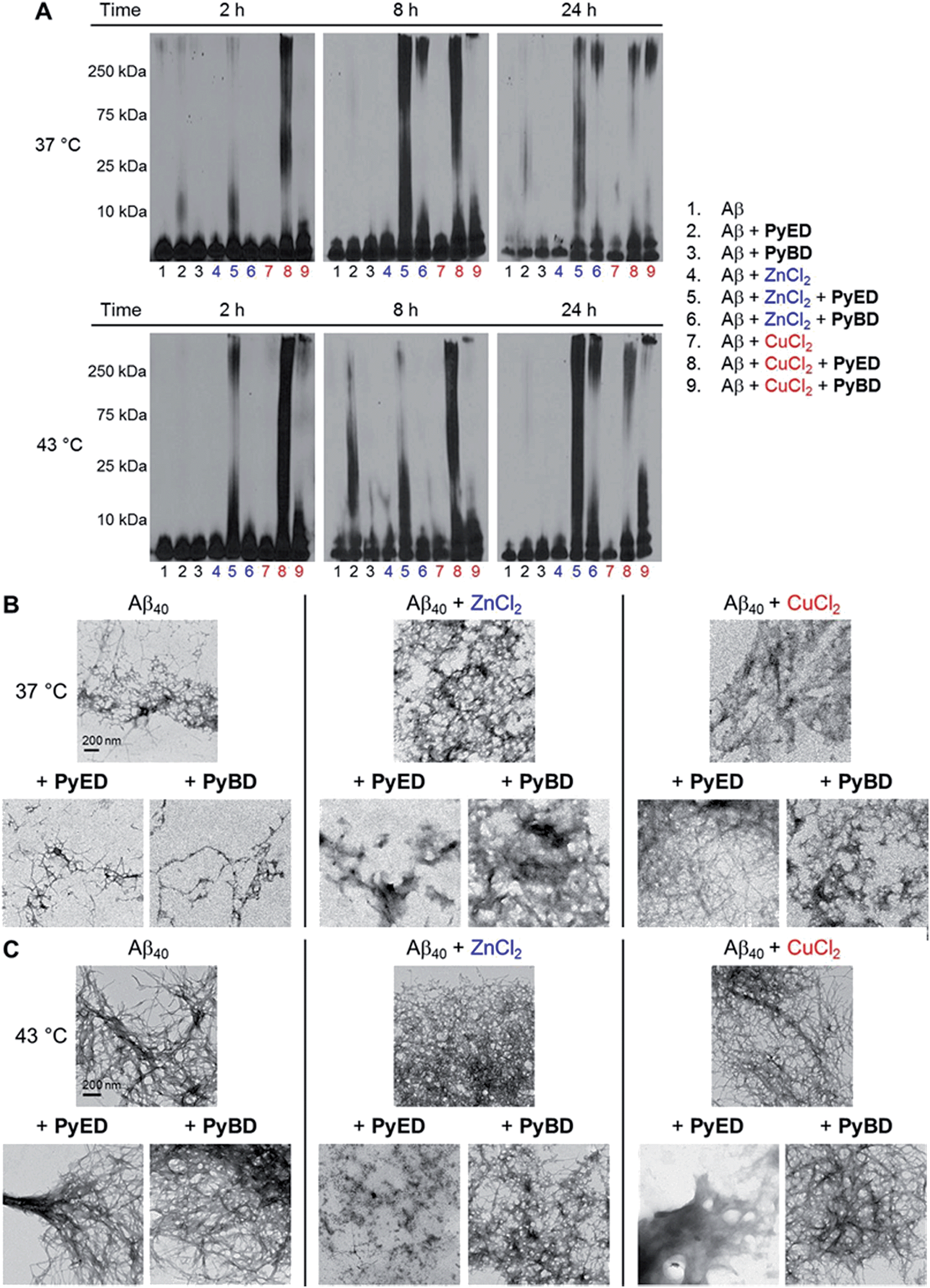 | ||
| Fig. 4 Reactivity of PyED and PyBD with preformed Aβ40 aggregates. (A) Analysis of resultant Aβ40 species by gel electrophoresis with Western blot using an anti-Aβ antibody (6E10). TEM images of the samples incubated for 24 h at (B) 37 °C or (C) 43 °C. Experimental conditions: [Aβ] = 25 μM; [CuCl2 or ZnCl2] = 25 μM; [PyED or PyBD] = 50 μM; 2, 8, 24 h incubation at 37 or 43 °C; pH 7.4 (metal-free and Zn(II)) or pH 6.6 (Cu(II)); constant agitation. | ||
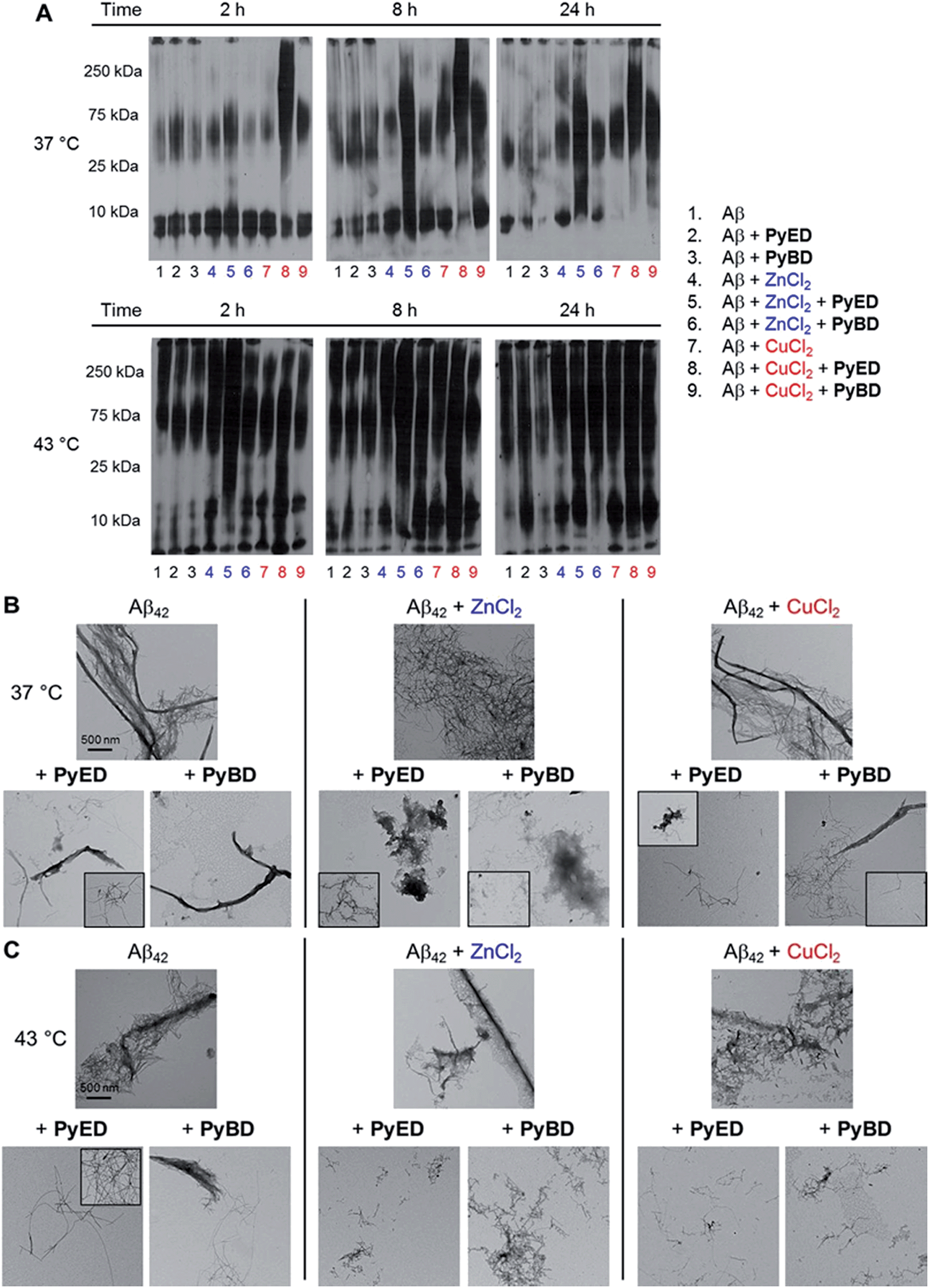 | ||
| Fig. 5 (A) Analysis of resultant Aβ42 species by gel electrophoresis with Western blot using an anti-Aβ antibody (6E10). TEM images of samples incubated for 24 h at (B) 37 °C or (C) 43 °C. Experimental conditions: [Aβ] = 25 μM; [CuCl2 or ZnCl2] = 25 μM; [PyED or PyBD] = 50 μM; 2, 8, 24 h incubation at 37 or 43 °C; pH 7.4 (metal-free and Zn(II)) or pH 6.6 (Cu(II)); constant agitation. | ||
For preformed metal-free and metal-associated Aβ40 and Aβ42 aggregates, PyED and PyBD exhibit differing disaggregation capabilities (Fig. 4 and 5). In the case of Zn(II)– and Cu(II)–Aβ40 samples treated with PyED, Aβ species with an increasing range of molecular weights (MW) are observed at both 37 and 43 °C between 2 and 8 h, while a decrease in signal intensity occurs between 8 and 24 h for samples incubated at 37 °C (Fig. 4A, lanes 5 and 8). Interestingly, variable reactivity of PyED with preformed Zn(II)–Aβ40 aggregates was detected at 43 °C. Addition of PyED presented an increasing distribution of MW throughout the time course (Fig. 4A, lane 5). In contrast, treatment of metal–Aβ40 samples with PyBD leads to the generation of lower MW species (MW ≤ 25 kDa) over the 24 h period (Fig. 4A, lanes 6 and 9). These data suggest that PyBD only slightly affects the transformation of preformed metal–Aβ aggregates, indicating that the introduction of radical formation upon metal binding by PyED may be a key factor in the generation of metal-associated Aβ species exhibiting a different array of MW. Additionally, the reduction of gel band intensities in Cu(II)–Aβ40 samples incubated with PyED for 24 h may imply the occurrence of further aggregation over time. These results are markedly different than those observed for metal-free samples incubated with PyED or PyBD which demonstrate overall minimal disaggregation activity, with the exception of 8 h incubation under hyperthermic conditions. This modest metal-free activity is expected due to the absence of both chelation and chelation-induced radical generation pathways (Fig. 4A, lane 2). In the absence of divalent metals, initiation of thermally-induced PyED cyclization is slow,64 leading to limited radical formation under these conditions.
The trends in the gel analysis are consistent with TEM images of preformed metal–Aβ40 aggregates treated with PyED. At 37 °C, TEM images of metal–Aβ40 show a mixture of fibrillar and amorphous structure types, while at 43 °C amorphous Aβ morphologies are dominant. In comparison, parallel samples incubated with PyBD exhibit fibrillar structures similar to those under compound-free conditions at both temperatures (Fig. 4B and C). Since PyED and PyBD show minimal change in the morphology of metal-free Aβ40 aggregates relative to untreated samples, this suggests that variations in the fibrillar morphology may derive from chelation and chelation-induced radical mechanisms (Fig. 4B and C).
The ability of PyED and PyBD to transform preformed Aβ42 aggregates was also examined (Fig. 5). Relative to analogous Aβ40 samples, a similar trend in both PyED and PyBD reactivity with Aβ42 was confirmed by gel electrophoresis. Aβ42 species with a wide distribution of MW are observed with PyED-treated, metal-associated Aβ42 aggregates over the course of 24 h at both temperatures (Fig. 5A, lanes 5 and 8), while low reactivity is visualized in metal–Aβ42 samples incubated with PyBD (lanes 6 and 9). In the case of metal-free conditions, a slightly different ensemble of Aβ42 MW are produced relative to the untreated control upon addition of PyED or PyBD (lanes 1–3). In addition to these trends, the TEM images reveal that analogous to Aβ40, addition of PyED to metal–Aβ42 samples induces changes in the morphology of preformed aggregates. Metal-treated Aβ42 exposed to either PyED or PyBD show thinner fibrils of various lengths (37 and 43 °C), as well as more amorphous species (37 °C) than observed in compound-free samples (Fig. 5B and C). As demonstrated for Aβ40, no distinct morphology changes are observed in the metal-free Aβ42 samples treated with either ligand when compared to the untreated sample, indicating the importance of metal chelation in the disaggregation pathway.
In an effort to evaluate whether chelation and radical generation can influence fibril assembly, the effect of PyED and PyBD on modulation of the Aβ aggregation pathway was investigated (Fig. S3 and S4†). Upon incubation of Zn(II) and Aβ40 with PyED, an increasing dispersion of various MW was visualized by gel analysis over prolonged exposures of up to 24 h (Fig. S3,† lane 5). In comparison, samples containing Cu(II), Aβ40, and PyED generate different smearing patterns than those of the analogous Zn(II)–Aβ40 samples (lanes 5 and 8). Similar to the disaggregation results, the gel band intensities of the Cu(II)–Aβ40 samples also decrease between 8 and 24 h incubation time at both 37 and 43 °C, suggesting the possibility of further aggregation over long incubation times. The PyED inhibition activity compares favorably with that of PyBD, where only a slight modulation of the metal-induced aggregation pathway is observed (lanes 6 and 9), while exposure of metal-free Aβ40 to either PyED or PyBD results in little to no activity (lanes 2 and 3). Thus, analogous to the disaggregation results, these data indicate that bifunctional PyED exhibits greater inhibition of metal-induced Aβ aggregation compared to monofunctional PyBD. These findings are supported by TEM images of Aβ40 samples incubated at 43 °C that reveal smaller, amorphous Aβ species in the presence of divalent metal ions and PyED (Fig. S3C†). Importantly, no significant morphological changes are observed in the metal-free Aβ samples exposed to either PyED or PyBD. As expected, the trend in the inhibition of Aβ42 aggregation upon addition of PyED or PyBD is comparable to that of Aβ40 (Fig. S4†). Significant modification of the aggregation pathway is only visualized for metal-associated Aβ species treated with PyED (Fig. S4A,† lanes 5 and 8). Additionally, TEM images reveal that thinner fibrils and/or amorphous aggregates are generated in PyED-treated metal–Aβ42 samples compared to compound-free conditions (Fig. S4B and C†). Taken together, the disaggregation and inhibition results reveal the enhanced ability of PyED (vs.PyBD) to variably modulate metal-free and metal-induced Aβ aggregation.
From the data available on a range of molecular structures, modulation of Aβ species may derive from the differential interaction between the ligand frameworks and monomeric Aβ peptide.4,30,32 To evaluate the degree of this interaction, Aβ was incubated with PyED or control ligand PyBD at 0 °C for 2 h in a ratio of 6:1 ligand:Aβ peptide ([Aβ] = 100 μM).30 The resulting species were analyzed using native nanoelectrospray ionization-mass spectrometry by comparison to the established Aβ-interacting neuropeptide Leucine-enkephalin (Leu-enk).90 No ligand–Aβ species were detected and no significant differences were observed between the spectra obtained upon incubation with PyED or PyBD, suggesting neither ligand framework appreciably binds Aβ relative to Leu-enk.
Overall, the data suggest that the disparate capability of PyED to regulate Aβ aggregation is due to a combination of metal extraction from Aβ, interaction of ligand with metal-bound Aβ species, and induction of radical-mediated modification of the peptide aggregation pathway. In support of the interaction of PyED and PyED with metal-bound Aβ species at 43 °C, photo-induced loss of HCO2 from M(II)–Aβ disaggregation samples and subsequent loss of CO2 (Cu(II)–Aβ only) on the b-ions is observed by MALDI-TOF-TOF, whereas the corresponding y-ions appear to be intact (Fig. S5 and Tables S1–S3†). Moreover, the b7 ion reveals that CO2 loss occurs N-terminal to aspartate, D7. In addition, MALDI-TOF data show that HCO2 loss is only operative upon incubation (4–8 h) of the components Aβ, metal ion (Cu(II) or Zn(II)), and ligand (PyBD or PyED) (Fig. S6–S8†). This loss of HCO2 from M(II)–Aβ and subsequent loss of CO2 (Cu(II)–Aβ only) is also observed in the analogous 37 °C disaggregation samples (Fig. S9–S11†). Importantly, rapid addition of any of the pair of components with the same incubation time, or the mixture of three components with no incubation does not result in any detectable photo-induced loss of CO2. This indicates that metal and ligand must be co-localized within the metal binding consensus sequence (1–16) and is consistent with CO2 loss mechanisms from photo-induced redox and ESI electron capture/detachment of Cu and Zn bound peptides.91–94
While evidence for the interaction among Aβ, metal ions, and PyED or PyBD is suggested for the Aβ modulation pathway, and radical-induced peptide fragmentation as part of the overarching Aβ degradation process is also proposed, detection of specific, low MW fragments is more elusive. Peptidic cleavage by α-H-atom abstraction and subsequent detection of fragments by mass spectrometry is complicated by solvent accessibility and side-chain reactivity which diversify product distribution and reduce the abundances of individual species. The absence of individual peptide fragments, however, does not preclude radical damage to Aβ as all of these radical reaction pathways will lead to structural changes in metal-bound Aβ aggregates. Thus, the enhanced reactivity of PyED compared to PyBD towards metal–Aβ species is reflective of the broader role radicals may play in the modulation of overall Aβ structure.
Conclusions
Approaches to modulate the aggregation pathway are at the forefront of current small molecule designs for AD therapy. The ensemble of existing methodologies encompasses metal chelation and interruption of peptide aggregation by ligand interaction with Aβ, as well as targeting metal–Aβ ternary complex formation as disruption mechanisms. Here we demonstrate a bifunctional approach that combines metal chelation and active radical generation to affect Aβ aggregation. Our results indicate that the ligand–metal–Aβ interaction with subsequent radical generation is a relatively rapid (2 h at 43 °C) mechanism for influencing Aβ structural integrity and thus, the aggregation pathway. This outcome may lead to new hybrid molecular constructs designed to take advantage of several Aβ interaction modes in order to achieve rapid Aβ species modulation.Acknowledgements
This work is supported by the National Science Foundation (CHE-1265703) (to J.M.Z.); the Ruth K. Broad Biomedical Foundation, the DGIST R&D Program of the Ministry of Science, ICT and Future Planning of Korea (14-BD-0403), and the National Research Foundation of Korea grant funded by the Korean government (MSIP) (NRF-2014R1A2A2A01004877) (to M.H.L.). The authors thank Joan M. Walker for acquiring the Aβ40 TEM images.Notes and references
- Alzheimer's Association, Alzheimers Dement., 2012, 8, 131–168 CrossRef PubMed
.
- F. M. LaFerla, K. N. Green and S. Oddo, Nat. Rev. Neurosci., 2007, 8, 499–509 CrossRef CAS PubMed
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed
- M. G. Savelieff, A.S. DeToma, J.S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed
- Y. Miller, B. Ma and R. Nussinov, Chem. Rev., 2010, 110, 4820–4838 CrossRef CAS PubMed
- K. Blennow, M. J. de Leon and H. Zetterberg, Lancet, 2006, 368, 387–403 CrossRef CAS
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS PubMed
- E. Karran, M. Mercken and B. De Strooper, Nat. Rev. Drug Discovery, 2011, 10, 698–712 CrossRef CAS PubMed
- M. F. Galindo, I. Ikuta, X. Zhu, G. Casadesus and J. Jordan, J. Neurochem., 2010, 114, 933–945 CAS
- J. P. Blass, Ann. N. Y. Acad. Sci., 2000, 924, 170–183 CrossRef CAS PubMed
- M. Dumont and M. F. Beal, Free Radical Biol. Med., 2011, 51, 1014–1026 CrossRef CAS PubMed
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS PubMed
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. d. Paradis, J.-P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464–1467 CAS
- A. I. Bush, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS PubMed
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2012, 120, 149–166 CrossRef CAS PubMed
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS PubMed
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed
- P. Faller, ChemBioChem, 2009, 10, 2837–2845 CrossRef CAS PubMed
- J. A. Duce and A. I. Bush, Prog. Neurobiol., 2010, 92, 1–18 CrossRef CAS PubMed
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC
- P. Zatta, D. Drago, S. Bolognin and S. L. Sensi, Trends Pharmacol. Sci., 2009, 30, 346–355 CrossRef CAS PubMed
- P. Faller and C. Hureau, Dalton Trans., 2009, 1080–1094 RSC
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y.-S. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS PubMed
- P. J. Crouch, D. J. Tew, T. Du, D. N. Nguyen, A. Caragounis, G. Filiz, R. E. Blake, I. A. Trounce, C. P. W. Soon, K. Laughton, K. A. Perez, Q.-X. Li, R. A. Cherny, C. L. Masters, K. J. Barnham and A. R. White, J. Neurochem., 2009, 108, 1198–1207 CrossRef CAS PubMed
- C. W. Ritchie, A. I. Bush and A. Mackinnon,
et al.
, Arch. Neurol., 2003, 60, 1685–1691 CrossRef PubMed
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2008, 7, 779–786 CrossRef CAS PubMed
- N. G. Faux, C. W. Ritchie, A. Gunn, A. Rembach, A. Tsatsanis, J. Bedo, J. Harrison, L. Lannfelt, K. Blennow, H. Zetterberg, M. Ingelsson, C. L. Masters, R. E. Tanzi, J. L. Cummings, C. M. Herd and A. I. Bush, J. Alzheimer's Dis., 2010, 20, 509–516 CAS
- A. S. Pithadia, A. Kochi, M. T. Soper, M. W. Beck, Y. Liu, S. Lee, A. S. DeToma, B. T. Ruotolo and M. H. Lim, Inorg. Chem., 2012, 51, 12959–12967 CrossRef CAS PubMed
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef
- J. J. Braymer, J.-S. Choi, A. S. DeToma, C. Wang, K. Nam, J. W. Kampf, A. Ramamoorthy and M. H. Lim, Inorg. Chem., 2011, 50, 10724–10734 CrossRef CAS PubMed
- Y. Liu, A. Kochi, A. S. Pithadia, S. Lee, Y. Nam, M. W. Beck, X. He, D. Lee and M. H. Lim, Inorg. Chem., 2013, 52, 8121–8130 CrossRef CAS PubMed
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS PubMed
- M. B. Goshe, Y. H. Chen and V. E. Anderson, Biochemistry, 2000, 39, 1761–1770 CrossRef CAS PubMed
- A. P. Breen and J. A. Murphy, Free Radical Biol. Med., 1995, 18, 1033–1077 CrossRef CAS PubMed
- L.-W. Wang and M. R. Chance, Anal. Chem., 2011, 83, 7234–7241 CrossRef CAS PubMed
- G. Xu and M. R. Chance, Chem. Rev., 2007, 107, 3514–3543 CrossRef CAS PubMed
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196–219 CrossRef CAS
- W. S. Bowen, W. E. Hill and J. S. Lodmell, Methods, 2001, 25, 344–350 CrossRef CAS PubMed
- H. Eguchi, Y. Ikeda, S. Koyota, K. Honke, K. Suzuki, J. M. C. Gutteridge and N. Taniguchi, J. Biochem., 2002, 131, 477–484 CrossRef CAS PubMed
- S. Basak and V. Nagaraja, Nucleic Acids Res., 2001, 29, E105 CrossRef CAS PubMed
- T. Kowalik-Jankowska, M. Ruta, K. Wisniewska, L. Lankiewicz and M. Dyba, J. Inorg. Biochem., 2004, 98, 940–950 CrossRef CAS PubMed
- E. Heyduk and T. Heyduk, Biochemistry, 1994, 33, 9643–9650 CrossRef CAS PubMed
- E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef CAS PubMed
- S. D. Maleknia and K. M. Downard, Chem. Soc. Rev., 2014, 43, 3244–3258 RSC
- C. L. Hawkins and M. J. Davies, J. Chem. Soc., Perkin Trans. 2, 1998, 2617–2622 RSC
- D. T. Sawyer, Coord. Chem. Rev., 1997, 165, 297–313 CrossRef CAS
- M. Strlic, J. Kolar, V.-S. Selih, D. Kocar and B. Pihlar, Acta Chim. Slov., 2003, 50, 619–632 CAS
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed
- J. A. Simpson, K. H. Cheeseman, S. E. Smith and R. T. Dean, Biochem. J., 1988, 254, 519–523 CrossRef CAS PubMed
- N. K. Urbanski and A. Beresewicz, Acta Biochim. Pol., 2000, 47, 951–962 CAS
- B. D. Schmidt and C. F. Meares, Biochemistry, 2002, 41, 4186–4192 CrossRef CAS PubMed
- K. B. Hall and R. O. Fox, Methods, 1999, 18, 78–84 CrossRef CAS PubMed
- M. A. Trakselis, S. C. Alley and F. T. Ishmael, Bioconjugate Chem., 2005, 16, 741–750 CrossRef CAS PubMed
- G. M. Heilek and H. F. Noller, Science, 1996, 272, 1659–1662 CAS
- L. Lancaster, M. C. Kiel, A. Kaji and H. F. Noller, Cell, 2002, 111, 129–140 CrossRef CAS PubMed
- P. A. MacFaul, D. D. M. Wayner and K. U. Ingold, Acc. Chem. Res., 1998, 31, 159–162 CrossRef CAS
- W. M. Garrison, Chem. Rev., 1987, 87, 381–398 CrossRef CAS
- R. T. Dean, S. P. Wolff and M. A. McElligott, Free Radical Res. Commun., 1989, 7, 97–103 CrossRef CAS PubMed
- M. J. Davies, Arch. Biochem. Biophys., 1996, 336, 163–172 CrossRef CAS PubMed
- E. R. Stadtman, Methods Enzymol., 1995, 258, 379–393 CAS
- C. J. Easton, Chem. Rev., 1997, 97, 53–82 CrossRef CAS PubMed
- D. S. Rawat and J. M. Zaleski, J. Am. Chem. Soc., 2001, 123, 9675–9676 CrossRef CAS PubMed
- T. Chandra, R. A. Allred, B. J. Kraft, L. M. Berreau and J. M. Zaleski, Inorg. Chem., 2004, 43, 411–420 CrossRef CAS PubMed
- S. E. Lindahl, H. Park, M. Pink and J. M. Zaleski, J. Am. Chem. Soc., 2013, 135, 3826–3833 CrossRef CAS PubMed
- P. J. Benites, R. C. Holmberg, D. S. Rawat, B. J. Kraft, L. J. Klein, D. G. Peters, H. H. Thorp and J. M. Zaleski, J. Am. Chem. Soc., 2003, 125, 6434–6446 CrossRef CAS PubMed
- B. J. Kraft, N. L. Coalter, M. Nath, A. E. Clark, A. R. Siedle, J. C. Huffman and J. M. Zaleski, Inorg. Chem., 2003, 42, 1663–1672 CrossRef CAS PubMed
- J. M. Walker, L. Gou, S. Bhattacharyya, S. E. Lindahl and J. M. Zaleski, Chem. Mater., 2011, 23, 5275–5281 CrossRef CAS
- S. M. Routt, J. Zhu, J. M. Zaleski and J. R. Dynlacht, Int. J. Hyperthermia, 2011, 27, 435–444 CrossRef CAS PubMed
- W. C. Dewey, S. A. Sapareto and D. A. Betten, Radiat. Res., 1978, 76, 48–59 CrossRef CAS PubMed
- P. M. Corry, S. Robinson and S. Getz, Radiology, 1977, 123, 475–482 CrossRef CAS PubMed
- E. Dikomey and J. Franzke, Int. J. Radiat. Biol., 1992, 61, 221–233 CrossRef CAS PubMed
- R. S. L. Wong, J. R. Dynlacht, B. Cedervall and W. C. Dewey, Int. J. Radiat. Biol., 1995, 68, 141–152 CrossRef CAS PubMed
- W. L. Titsworth, G. J. A. Murad, B. L. Hoh and M. Rahman, Anticancer Res., 2014, 34, 565–574 Search PubMed
- D. M. Welsh, Crit. Care Nurs. Clin., 1995, 7, 115–123 CAS
- M. H. Falk and R. D. Issels, Int. J. Hyperthermia, 2001, 17, 1–18 CrossRef CAS PubMed
- Y. Anzai, R. Lufkin, A. DeSalles, D. R. Hamilton, K. Farahani and K. L. Black, Am. J. Neuroradiol., 1995, 16, 39–48 CAS
- C. Rodriguez-Rodriguez, N. Sanchez de Groot, A. Rimola, A. Alvarez-Larena, V. Lloveras, J. Vidal-Gancedo, S. Ventura, J. Vendrell, M. Sodupe and P. Gonzalez-Duarte, J. Am. Chem. Soc., 2009, 131, 1436–1451 CrossRef CAS PubMed
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS PubMed
- A. Avdeef, S. Bendels, L. Di, B. Faller, M. Kansy, K. Sugano and Y. Yamauchi, J. Pharm. Sci., 2007, 96, 2893–2909 CrossRef CAS PubMed
- D. E. Clark and S. D. Pickett, Drug Discovery Today, 2000, 5, 49–58 CrossRef CAS PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed
- BBB Protocol and Test Compounds, pIon, Inc., Woburn, MA, 2009 Search PubMed.
- J. Masuoka, J. Hegenauer, B. R. Van Dyke and P. Saltman, J. Biol. Chem., 1993, 268, 21533–21537 CAS
- L. D. Pettit and J. L. M. Swash, J. Chem. Soc., Dalton Trans., 1976, 588–594 RSC
- H. Zhang, C.-S. Liu, X.-H. Bu and M. Yang, J. Inorg. Biochem., 2005, 99, 1119–1125 CrossRef CAS PubMed
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed
- M. T. Soper, A. S. DeToma, S.-J. Hyung, M. H. Lim and B. T. Ruotolo, Phys. Chem. Chem. Phys., 2013, 15, 8952–8961 RSC
- J. Coon, J. Shabanowitz, D. Hunt and J. P. Syka, J. Am. Soc. Mass Spectrom., 2005, 16, 880–882 CrossRef CAS PubMed
- E. Bagheri-Majdi, Y. Ke, G. Orlova, I. K. Chu, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. B, 2004, 108, 11170–11181 CrossRef CAS
- R. E. Bossio, R. R. Hudgins and A. G. Marshall, J. Phys. Chem. B, 2003, 107, 3284–3289 CrossRef CAS
- T.-Y. Huang, J. F. Emory, R. A. J. O'Hair and S. A. McLuckey, Anal. Chem., 2006, 78, 7387–7391 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and supplementary figures and tables. See DOI: 10.1039/c4sc01979b |
| This journal is © The Royal Society of Chemistry 2015 |