
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studying the active-site loop movement of the São Paolo metallo-β-lactamase-1†
Jürgen
Brem‡
a,
Weston B.
Struwe‡
b,
Anna M.
Rydzik
a,
Hanna
Tarhonskaya
a,
Inga
Pfeffer
a,
Emily
Flashman
a,
Sander S.
van Berkel
a,
James
Spencer
c,
Timothy D. W.
Claridge
a,
Michael A.
McDonough
a,
Justin L. P.
Benesch
*b and
Christopher J.
Schofield
*a
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk
bDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: justin.benesch@chem.ox.ac.uk
cSchool of Cellular and Molecular Medicine, University of Bristol, Medical Sciences Building, Bristol, BS8 1TD, UK
First published on 24th October 2014
Abstract
Metallo-β-lactamases (MBLs) catalyse the hydrolysis of almost all β-lactam antibiotics. We report biophysical and kinetic studies on the São Paulo MBL (SPM-1), which reveal its Zn(II) ion usage and mechanism as characteristic of the clinically important di-Zn(II) dependent B1 MBL subfamily. Biophysical analyses employing crystallography, dynamic 19F NMR and ion mobility mass spectrometry, however, reveal that SPM-1 possesses loop and mobile element regions characteristic of the B2 MBLs. These include a mobile α3 region which is important in catalysis and determining inhibitor selectivity. SPM-1 thus appears to be a hybrid B1/B2 MBL. The results have implications for MBL evolution and inhibitor design.
Introduction
The β-lactams (BLAs) remain the most important class of antibiotics, representing ∼60% of those used in the clinic.1 However, β-lactam antibiotic (BLA) utility is threatened by resistance mechanisms, most importantly those involving plasmid encoded β-lactamases (BLs).2 BLs can be divided into two mechanistic classes – the serine-BLs (SBLs) and the metallo-BLs (MBLs).2,3 Although for many decades the SBLs were the most clinically relevant BLs, global dissemination of MBLs, e.g. the New Delhi MBLs (NDMs) and the São Paulo MBL (SPM-1), is now occurring.4–6 This is concerning because, with the exception of the monobactams, MBLs hydrolyze all known β-lactam antibiotics (BLAs) (Fig. 1A), and, whilst there are clinically useful SBL inhibitors, there are no such compounds for MBLs.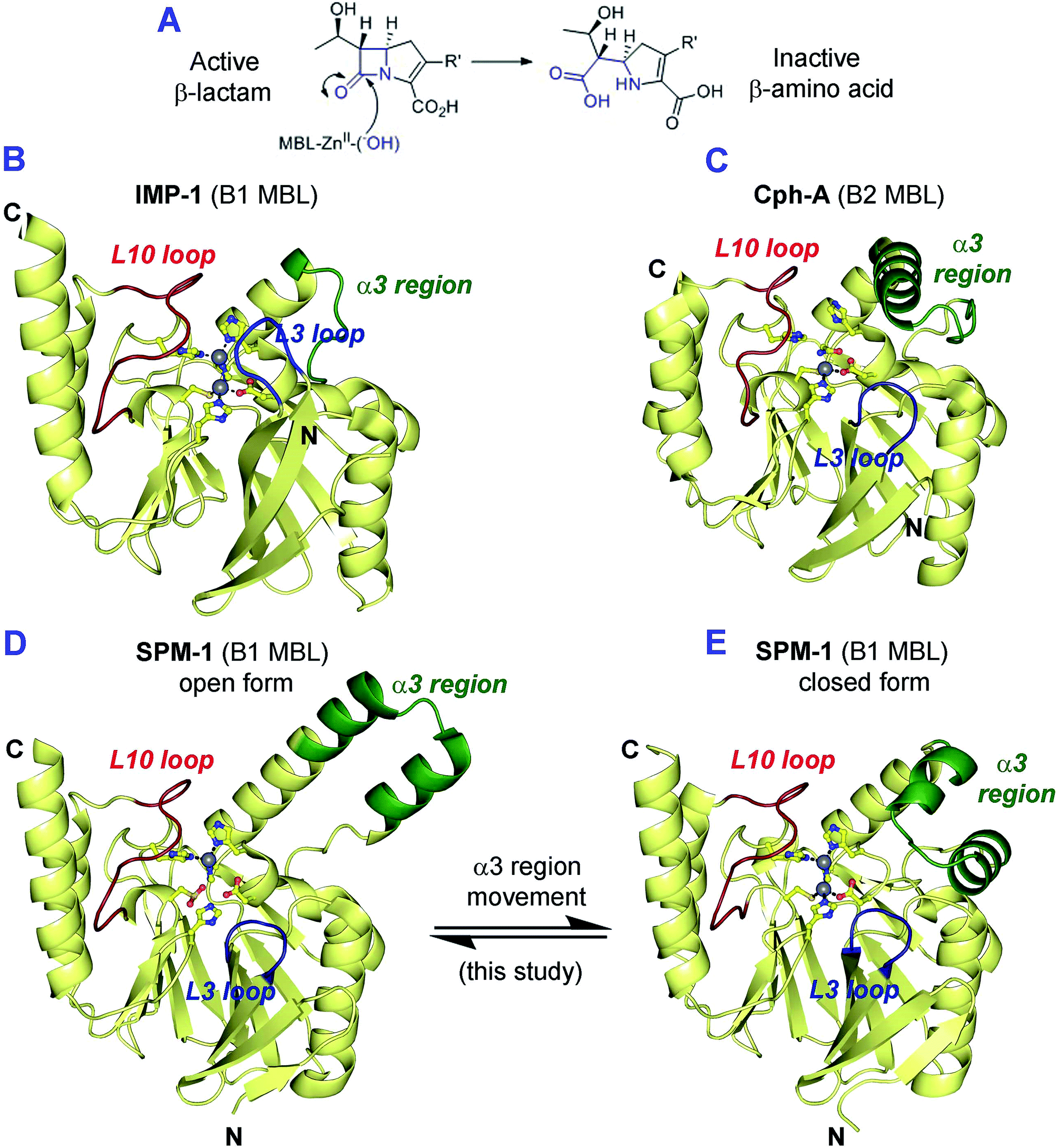 | ||
| Fig. 1 (A) Outline mode of action of metallo-β-lactamases (MBLs); (B–E) views from MBL crystal structures highlighting potentially mobile regions. Views of (B) IMP-1 (B1 MBL, PDB ID: 1JJT),20 (C) CphA (B2 MBL, PDB ID: 1X8I)17 and the (D) ‘open’ and (E) ‘closed’ forms of SPM-1 (B1 MBL, ‘open’ PDB ID: 2FHX14 and ‘closed’ PDB ID: 4BP0) highlighting the different mobile regions (L3 loop (blue), L10 loop (red) and α3 region (green)) that characterize the MBL subfamilies. A longer L3 loop (blue) is characteristic of the di-Zn B1 MBLs. The B2 MBLs using mono-zinc ion are characterised by an elongated α3 region (green) and a shorter L3 loop (blue). In the case of the B1 MBLs only SPM-1 has an elongated α3 loop (green) and a short L3 loop (blue). | ||
The MBL fold is structurally unrelated to those of SBLs. MBLs (class B of the Ambler β-lactamase classification)7 are divided into the B1, B2 and B3 subclasses reflecting sequence/structural relationships and substrate selectivities (Table S1, Fig. S1 and S2†)2 with the B1 and B2 MBLs being most closely related to each other. Most, but not all, clinically relevant MBLs are from subclass B1 (Table S1†).8
SPM-1 was first identified in a Pseudomonas aeruginosa strain from a paediatric leukaemia patient9 and is a major clinical problem in South America;10,11 in Brazil SPM-1 occurs in ∼35% of clinically identified carbapenem resistant P. aeruginosa isolates and is a major cause of infection mediated mortality.12 Unusually, SPM-1 has high sequence similarity with MBLs from both B1 (35% identity with IMP-1 and 30% with BcII) and B2 (32% with ImiS and CphA) MBL subclasses.13,14 Studies on MBL Zn(II) ion use reveal that the B1 MBLs use two zinc ions for efficient catalysis, whereas the B2 MBLs are inhibited by binding of a second Zn(II) ion (Fig. 1 and Table S1†).8 Interestingly, SPM-1 is reported to have a Zn(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MBL ratio of 1.5:1 in solution (by atomic absorption spectrometry); however, the single reported SPM-1 structure reveals only one zinc ion, bound at the tri-histidine or Zn1 site.13
:MBL ratio of 1.5:1 in solution (by atomic absorption spectrometry); however, the single reported SPM-1 structure reveals only one zinc ion, bound at the tri-histidine or Zn1 site.13
Structural studies on MBLs have revealed the importance of mobile regions in catalysis and substrate selectivity.8 Understanding how these loops influence binding to MBL active sites is important with respect to identifying MBL inhibitors with a sufficiently broad spectrum of activity for clinical utility. The different MBL subfamilies are distinguished by their use of different loops/mobile regions for interactions with substrates (Table S1†). B1 MBLs are characterised by the presence of a L3 loop, located between strands β3 and β4 (residues 61–66, using standard numbering scheme for the class B β-lactamases (BBL numbering)),15 and a L10 loop (residues 223–241 (BBL numbering)), located between strand β11 and the helix α4, which includes the Arg/Lys224 and Asn233 residues that are involved in substrate binding.16 The B2 MBLs also have a L10 loop, but feature a shorter L3 loop that forms a tight turn between β3 and β4, and an extended region adjacent to the active site (‘α3 region’) that compromises the N-terminal section of a ‘kinked’ α-helix (α3), followed by a loop17 (Fig. 1 and S1–S4†).
In all reported B2 MBL crystal structures the α3 region has been observed in a ‘closed’ conformation, enabling the α3 loop to form a ‘hydrophobic wall’18 that defines the active site and contributes to binding of carbapenem substrates.14,17 However, EPR studies imply that β-lactam hydrolysis by B2 MBLs involves conformational changes that may be associated with their α3 regions.19
SPM-1 is unique amongst known B1 MBLs because it contains an extended α3 region (24 residues), and a relatively short L3 loop, both features characteristic of B2 enzymes (Fig. 1, S1 and S3–S4†).9,13,14 However, the only reported SPM-1 crystal structure (PDB ID: 2FHX; 1.9 Å resolution; space group R3) reveals the SPM-1 α3 region in an apparently ‘open’ conformation in which the α3 kink is not evident; instead this region forms an extended helix rendering the α3 loop sequence ∼30 Å from the active site.14 The SPM-1 α3 region also contains an additional α-helix (α4) that is not present in known B2 MBLs.14 Despite these apparent structural differences, deletion of the α3 region in SPM-1 causes a substantial decrease in catalytic activity against carbapenems,14 implicating this region in binding and/or hydrolysis of substrates. SPM-1 thus uniquely presents structural/sequence similarities to both B1 and B2 subclasses21 and in consequence is challenging from the perspective of developing broad spectrum MBL inhibitors.
Here we report solution and crystallographic studies of the role of the α3 region in the binding and hydrolysis of β-lactams by SPM-1, and in its interactions with inhibitors.
Results and discussion
To investigate whether the conformational flexibility of the SPM-1 α3 region is involved in substrate binding as was observed for B2 MBLs,19 we performed crystallisation experiments to obtain an alternative SPM-1 structure (Fig. S5, S7 and S8†). A resultant SPM-1 structure, (2.2 Å resolution; space group P212121, Table S2†) reveals a profound rearrangement of the α3 region resulting in a ‘closed’ form of the enzyme reminiscent of a B2 MBL. The new structure reveals a ‘closed’ α3 region with a kink in helix α3 that is very similar to B2 MBLs (Fig. 1). With the exception of the α3 region (Cα atoms of residues, 141–166, mean RMSD ∼ 4.0 Å), the remainder of the SPM-1 αβ/βα core fold that defines the MBL superfamily enzymes is very similar to the ‘open’ SPM-1 structure (all backbone Cα atoms minus the α3 loop region RMSD 0.29 Å).In addition to the ‘closed’ conformation of the α3 region, the new SPM-1 structure differs from that previously reported in that both zinc binding sites are fully occupied (Fig. 2B) (with the tri-histidine (Zn1) and Cys-His-Asp (Zn2) sites connected by a “bridging” water/hydroxide molecule with four- and five-fold coordination, respectively).
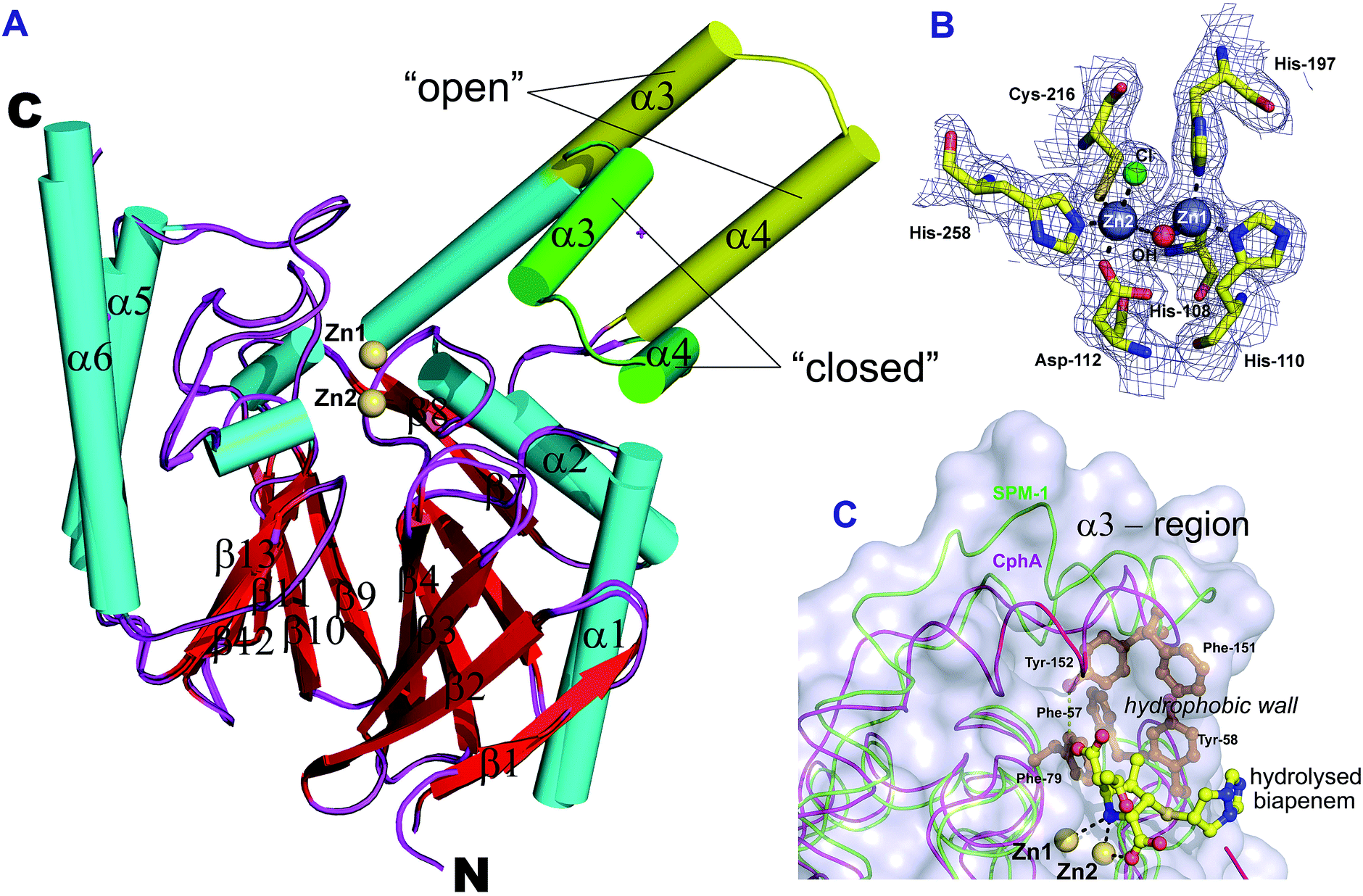 | ||
| Fig. 2 Crystallographic evidence for conformational changes in SPM-1. (A) Comparison of SPM-1 structures with the α3 region in an ‘open’ (PDB ID: 2FHX) and ‘closed’ (PDB ID: 4BP0) conformation. (B) Representative 2Fo–Fc electron density contoured to 1σ (blue mesh) of the active site residues and Zn atoms for the ‘closed’ SPM-1 structure. (C) Superimposition of CphA (purple, PDB ID: 1X8I) and the ‘closed’ SPM-1 structure (green), showing the position of hydrolysed biapenem in the active site. | ||
This observation is consistent with the presence of two Zn(II) ion equivalents in the purified recombinant protein, as measured by non-denaturing mass spectrometry (Fig. S7C†). The difference in the Zn(II):SPM-1 ratio that we observed (i.e. 2:1), compared to that previously reported (1:1),14 likely reflects (partial) oxidation of the active site Cys-residue (Cys221) that was observed in the ‘open’ conformation SPM-1 structure.14 Such oxidation has been shown to be linked to inhibition of metal binding to B1 MBLs.22 Thus, at least with respect to Zn(II)-binding characteristics, SPM-1 appears to behave as a typical B1 MBL (see below for solution studies). The positions of the α3 helix and α3 loop in the ‘closed’ SPM-1 structure are strikingly similar to those observed for the α3 region in the B2 MBL, CphA (Fig. 2C, 1 and S3†). As for CphA,17 the ‘closed’ conformation enables SPM-1 to form a hydrophobic wall, as observed for B2 MBLs, formed by residues Ala148, Phe151, Tyr152 and Leu157 (from the α3 loop) and Phe57 (β2), Tyr58 (β3) and Phe79 (from the loop linking β5 and α1) (Fig. 2C). However, the closed SPM-1 structure differs from the CphA, and other B2 structures, in that α3 is extended by 7 residues and by the presence of the α4 helix.
In contrast with the α3 helix, the α4 helix not only flanks the active site of the enzyme but also provides direct contact with it. In particular, the salt bridge between Arg160 (α4), Asp56 (short L3 loop) and Glu80 (α1) is notable. In addition, the SPM-1 active site is notably wider than those of the B2 MBLs (Fig. S3†), possibly contributing to the broader substrate specificity of SPM-1 (which hydrolyzes all β-lactams except monobactams)13 compared to the B2 MBLs (which are narrow-spectrum carbapenemases).
In order to examine whether SPM-1 undergoes conformational changes involving the α3 region on ligand binding, as proposed for B2 MBLs19 and implied by the SPM-1 crystal structures, we used non-denaturing ion mobility mass spectrometry (IM-MS) to investigate the conformational flexibility of SPM-1. IM-MS is based on the separation (or drift time) of a particular protein ion in a neutral gas (He) under the influence of a weak electric field. The drift time of a gas-phase ion is related to its rotationally averaged collisional cross section (CCS) which is specific to each protein charge state.23,24
IM-MS was used to investigate different conformational states of SPM-1 with and without Zn bound through reporting their corresponding CCS values. An IM-MS spectrum under conditions that preserve non-covalent interaction shows three major charge states (9+ → 11+) of SPM-1 with distributions of z = 9+ (10%), 10+ (60%), 11+ (30%), corresponding to a mass of 28030.5 Da (Fig. 2A). The CCS of the charge states are: 2119 Å2 (9+), 2167 Å2 (10+) and 2247 Å2 (11+) (Fig. 3B). The calculated CCS from X-ray structures of ‘open’ and ‘closed’ forms of SPM-1 are 2237 Å2 (open) and 2171 Å2 (closed). The ‘open’ crystal CCS thus correlates with the CCS of the 11+ SPM-1 ion and the ‘closed’ form matched the CCS of the 10+ charge state, suggesting the presence of both forms.
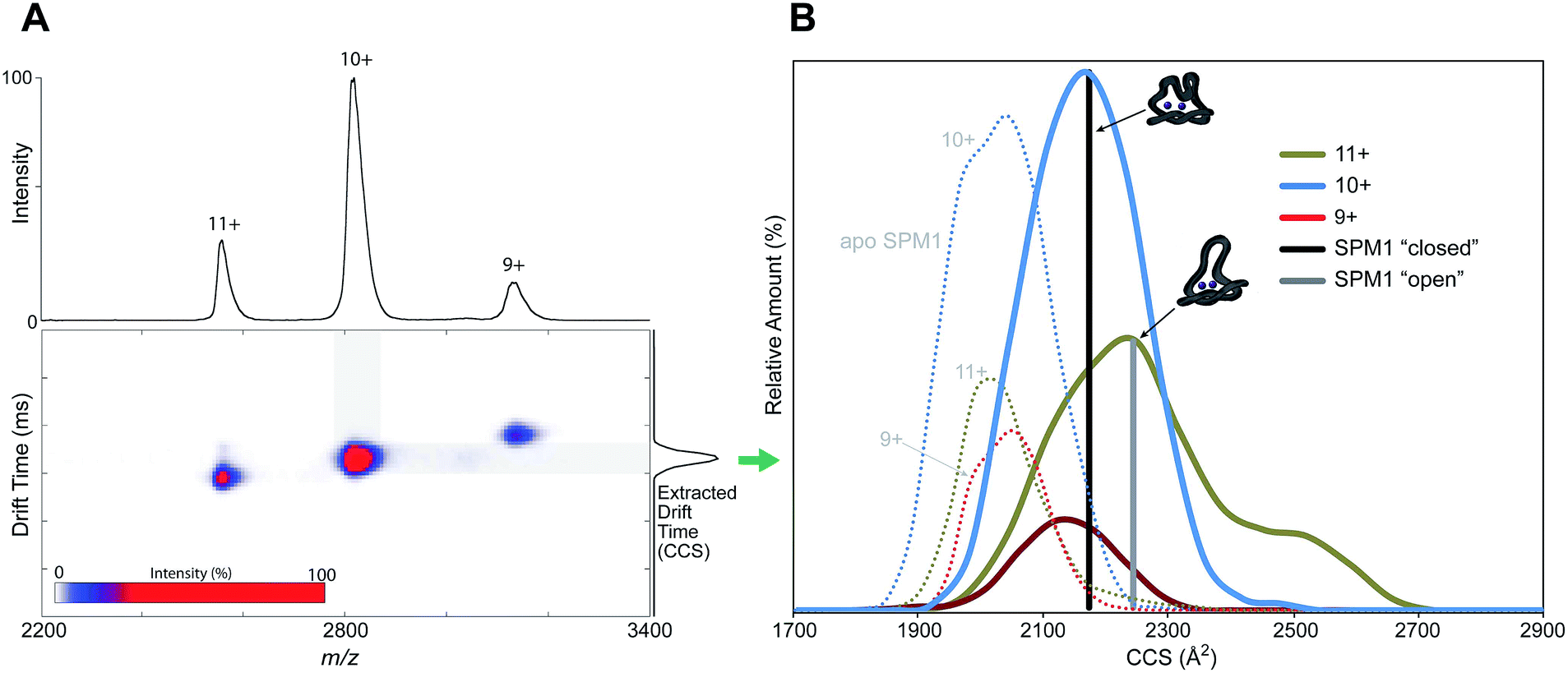 | ||
| Fig. 3 Non-denaturing ion mobility MS reveals different conformations of di-Zn(II)-SPM-1 as compared to the theoretical collisional cross section (CCS) values of the ‘open’ and ‘closed’ crystal forms. | ||
CCS measurements of apo-SPM-1, generated by EDTA treatment (Fig. 3B and S9†), showed a more compact conformation than the ‘closed’ form. The experimentally determined values for apo-SPM-1 are (2050 Å2 (9+), 2043 Å2 (10+), 2032 Å2 (11+)) – the difference from the di-Zn(II)-SPM-1 likely reflect changes in conformation apparent in the gas phase due to metal binding. In contrast to SPM-1, IM-MS measurements for di-Zn(II)-IMP-1, the B1 MBL with the highest sequence similarity to SPM-1 (35% identity), shows minimal differences in the CCSs of the 3 observed charge states, i.e. 2023 Å2 (10+), 1991 (9+), 1994 (8+), with a standard deviation of 17.5%, suggesting reduced loop flexibility for the di-Zn(II)-IMP-1 (Fig. S9†), compared to SPM-1, consistent with the relatively large α3 region in di-Zn(II)-SPM-1 compared to IMP-1.
To investigate whether the conformational flexibility observed by IM-MS is related to the α3 loop movement in solution, we then employed 19F NMR, which has been reported to be useful for monitoring loop movements in MBLs.25 To introduce a 19F label, Tyr152 at the C-terminal end of the α3 helix was substituted for a cysteine (Y152C) (Fig. 4, S10 and 11†); the Y152C variant was then selectively alkylated using 3-bromo-1,1,1-trifluoroacetone (BFA) to give the desired 19F-labeled di-Zn(II)-SPM-1 variant (SPM-1*), as verified by MS analyses (Fig. S13 and S14†).
In the 19F-NMR spectrum of SPM-1*, distinct signals at δ −72 ppm, and δ −83 ppm, respectively (in a 2:3 ratio, Fig. S17†) were observed, indicating that the BFA label is sampling two distinct environments and consistent with the presence of two conformational species which are in slow exchange on the NMR timescale (Fig. 4B). Denaturation with 2 M guanidine resulted in observation of two species at δ −84 ppm (Fig. 4A and S15†), supporting the proposal that the two signals observed with folded protein correspond to different conformations, note an analogous shift at δ −84 ppm was observed for denatured M67C NDM-1, Fig. S15.†
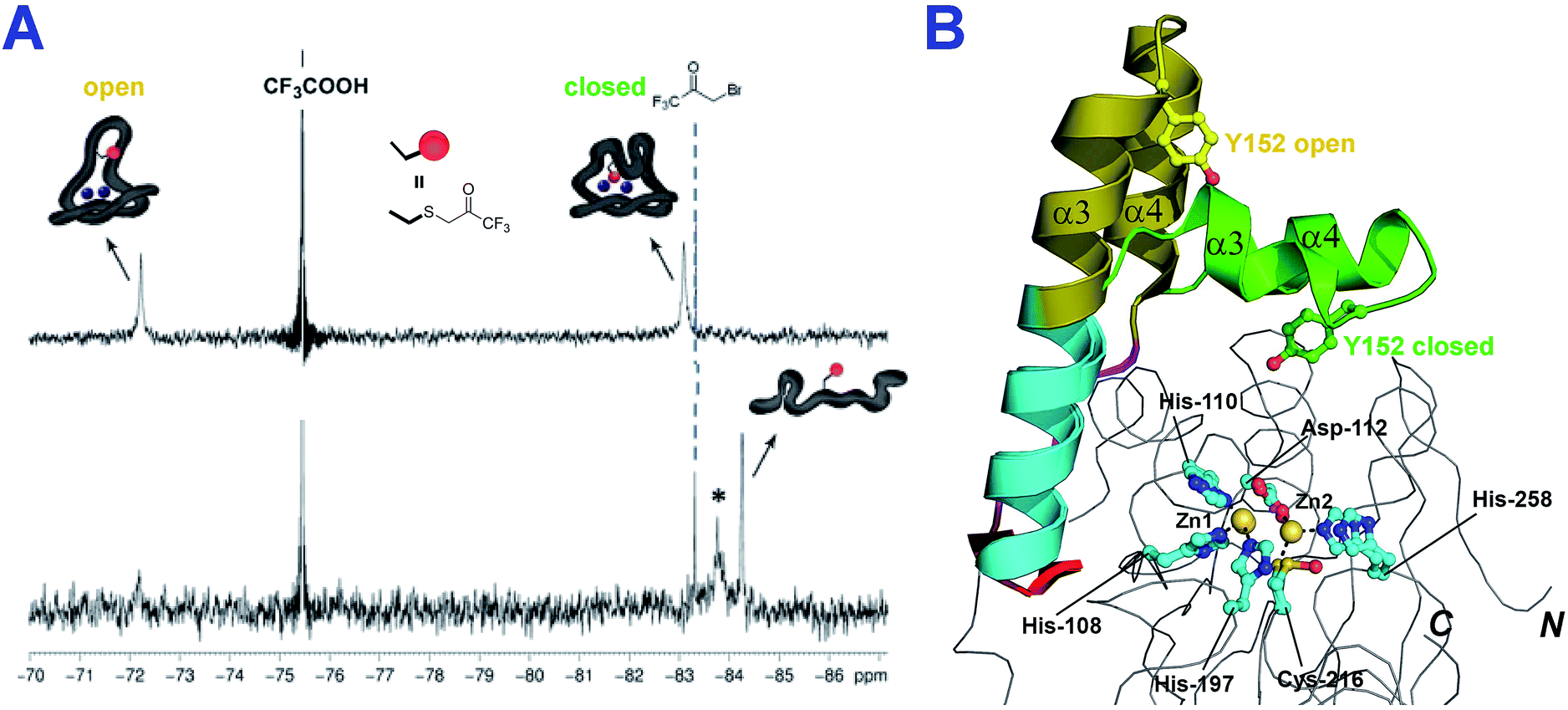 | ||
| Fig. 4 The loop of SPM-1 adopts distinct conformations in solution. (A) 19F-NMR spectra of folded di-Zn(II)-SPM-1* (top) and denatured SPM-1* (bottom) revealed distinct signals (The peak labeled * may correspond to an intermediate state between non-denatured and denatured states); (B) views of the SPM-1 α3 region in ‘closed’ and ‘open’ conformations, showing the different position of Tyr-152 (the residue which was substituted for Cys, derivatized with CF3COCH2Br, to give a Enz–SCH2COCF3 species). | ||
Removal of both Zn(II) ions from SPM-1* using 5 mM EDTA, led to the observation of distinct signals at δ −72 ppm, and δ −83 ppm respectively, (in a 1:4 ratio, Fig. S16 and 17†) as observed for the di-Zn(II)-SPM-1*, indicating the presence of two conformational species for apo-SPM-1* in solution. Note, this observation contrasts with the IM-MS results, where two conformations were observed for di-Zn(II)-SPM-1, but only one for apo-SPM-1, possibly due to the overall more compact nature of the latter, as analysed by IM-MS.
Investigation of solvent accessibility of the 19F label reveals the δ −72 ppm fluorine of SPM-1* as being much more solvent accessible (99% compared to CF3COOH standard) than the δ −83 ppm fluorine (58% compared to CF3COOH), suggesting that the δ −83 ppm peak corresponds to the closed form of SPM-1 (Fig. S18†.).
19F NMR saturation transfer experiments were then conducted; the results are consistent with the existence of one species (SPM-1*) in two different conformational states (assigned as ‘open’ and ‘closed’) (Fig. S19 and Table S3†). The saturation transfer results reveal that the exchange between the two states is slow on the NMR timescale (Table S3†). At 298 K a small decrease in signal intensity arising from chemical exchange was observed (δ −72 ppm, 28%; δ −83 ppm, 5%); at 320 K a larger decrease was observed (δ −72 ppm, 70%; δ −83 ppm, 25%), consistent with increased mobility at higher temperature.
Following the initial observation of exchange between the two fluorine resonances of SPM-1* by direct saturation transfer, the exchange rate was determined through 1D selective magnetization transfer experiments as appropriate for slow exchange systems;26–28 for significant exchange to be observed it was necessary to warm the sample to 310 K. Selective inversion of one resonance was followed by a variable mixing time in which exchange between the two sites could occur (in addition to longitudinal spin relaxation), after which a read pulse produced the 1D spectrum. The resulting magnetization exchange data are summarized in Table S5† and shown in Fig. 5. The selective magnetization transfer experiments reveal an exchange rate constant of 0.6 ± 0.1 s−1.
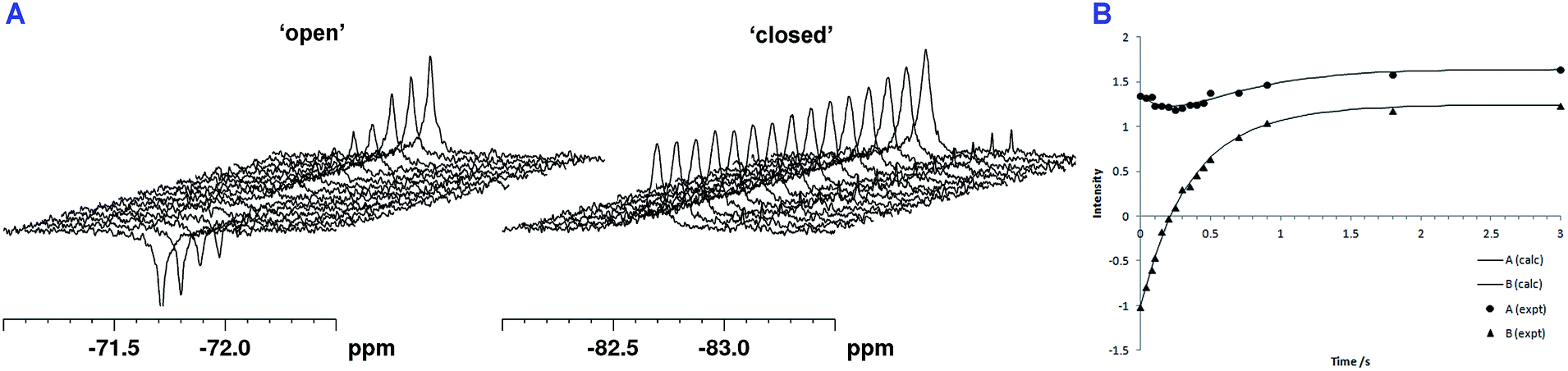 | ||
| Fig. 5 1D selective magnetization transfer experiments recorded at 310 K enable the exchange rate of loop movement to be determined. (A) 1D 19F exchange NMR spectra. (B) Fitted exchange data for SPM-1* (peak A = −82.2 ppm, peak B = −71.4 ppm). The solid curves are derived from the CIFIT fits. | ||
The combined X-ray crystallographic, ion mobility mass spectrometry and 19F NMR experiments, thus reveal that the α3 region of SPM-1 can adopt a conformation where it structurally resembles its equivalent in B2 MBLs in both the crystalline and solution states. This conclusion supports the proposal that, despite possession of a binuclear zinc center typical of B1 MBLs, in structural respects SPM-1 more closely resembles the B2 rather than the B1 MBLs.
To further test this hypothesis we investigated inhibition of SPM-1 with compounds of known selectivity for B1 and B2 MBL subclasses. We found that L-(S,S)-captopril, which is an established B1 MBL subclass inhibitor,29 but which does not potently inhibit the B2 MBL CphA,30 does not inhibit SPM-1 at concentrations up to 100 μM. Furthermore, 3,5-bis(mercaptomethyl)benzoic acid, which is a more potent inhibitor of B2 MBLs (CphA IC50 = 0.09 μM)30 than B1 MBLs, inhibits SPM-1 with a sub-micromolar potency (IC50 0.7 μM).3119F NMR revealed that whilst L-(S,S)-captopril does not bind to SPM-1* within limits of detection (at 1 mM), 3,5-bis(mercaptomethyl)benzoic acid and another thiol inhibitor (thiosalicylic acid, IC50 > 100 μM) cause line broadening and changes in 19F chemical shift (Fig. S20 and S21†). These results imply that the inhibition profile of SPM-1 may be most characteristic of B2 MBLs, and indicate involvement of the α3 region in inhibitor binding.
We then carried out kinetic studies of β-lactam hydrolysis by SPM-1, with the aim of comparing the results with previously characterised B1 and B2 MBLs, which are proposed to operate via different mechanisms.8 Hydrolysis of the fluorogenic cephalosporin substrate FC532 (6R,7R)-8-oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 5,5-dioxide; (2.5 μM substrate and 0.5 nM enzyme, pH = 7.5) reveals an increase in activity with increasing Zn(II) concentration up to 500 μM (Fig. S22†), consistent with the behaviour of B1 MBLs such as BcII,33 rather than of B2 enzymes that are generally inhibited by additional Zn(II) equivalents.8
We used stopped-flow analyses to investigate hydrolysis of the carbapenem antibiotic meropenem by either di-Zn(II) or Co(II) substituted SPM-1. Exposure of meropenem to di-Zn(II) SPM-1 generated a transiently populated species absorbing at 380 nm (Fig. 6A), which appeared within the mixing time of the experiment and disappeared within 500 ms. A similar signal was observed when imipenem is hydrolysed by di-Zn(II) BcII.34 To investigate this further, Co(II)-substituted SPM-1 was used, enabling us to exploit the spectroscopically ‘active’ Co(II) to obtain additional information on events taking place within the active site during meropenem turnover. Previous studies with BcII further suggest that the reduction in activity of the Co(II)-substituted, compared to the Zn(II) enzyme permits both accumulation and loss of intermediate species to be observed.34 Co(II)-substituted SPM-1 was derived by exposure of apo-SPM-1, generated by EDTA treatment as above, to Co(II) at a range of concentrations. The resulting protein possesses an absorption spectrum featuring both a prominent band at 350 nm (characteristic of a Cys → Co(II) ligand-metal charge transfer (LMCT) band) and a series of less intense features at visible wavelengths (between 500 and 650 nm) that may be attributed to ligand:field transitions. The spectrum is reminiscent of that reported for Co(II)-substituted BcII33 and can be distinguished from that of the Co(II)-substituted B2 MBL ImiS35 by the presence of additional features in the ligand:field region.
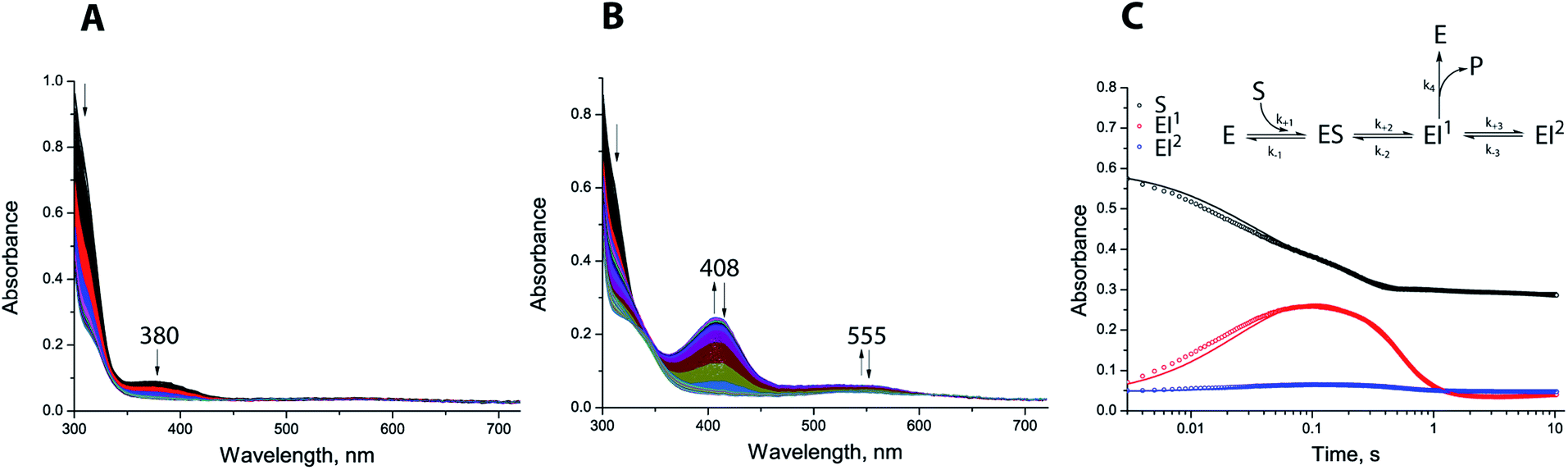 | ||
| Fig. 6 Stopped-flow analysis of SPM-1 catalysed meropenem hydrolysis. (A) Apo-SPM-1 (100 μM) was incubated with Zn(II) (200 μM) for ∼10 min then mixed with meropenem (100 μM) (5 °C); (B) apo-SPM-1 (100 μM) was incubated with Co(II) (500 μM) for ∼10 min, then mixed with meropenem (100 μM); (C) global fit traces and proposed kinetic mechanism. Global fit traces and proposed kinetic mechanism (assuming that only di-Co(II) substituted SPM-1 (E) is present in solution before the reaction due to 5-fold excess of Co(II) used for the experiment). | ||
Co(II)-substituted SPM-1 was next analysed using stoichiometric meropenem as a substrate at a range of Co(II) concentrations (1:0.5, 1:1, 1:2 and 1:5 enzyme:Co(II) ratios) (Fig. 6, S23–S26 and Table S5†). Following initiation of the reaction with meropenem by rapid mixing, we observed transient appearance of a relatively strong absorbance band at 408 nm and a more weakly absorbing feature at 555 nm (Fig. 6B). We propose these observations are analogous to the absorbance bands at 407 nm and 575 nm reported for the reaction of Co(II) complexes of the B1 MBL BcII with imipenem and meropenem.34 Accumulation of the intermediate species was dependent on Co(II) concentration and reached a maximum at a SPM-1:Co(II) ratio of 1:2 (Fig. S23–S26 and Table S5†), implicating the di-Co(II) species in stabilisation of the putative intermediate(s). Traces at 310, 408 and 555 nm were then fitted to evaluate the ability of a range of possible kinetic schemes to describe the experimental observations (Tables S5 and S6†). The fits utilised data collected at excess Co(II) (1:5 SPM-1:Co(II) ratio), simplifying the analysis by permitting the assumption that the reaction mixture contains only di-Co(II) SPM-1 and that no other enzyme species is contributing to the reaction.
The best fitting model (Fig. 6C and S26†) involves a branched mechanism where a populated intermediate EI1, distinguished by strong absorbance at 408 nm, can both yield product and equilibrate with an unproductive species EI2. Such a branched mechanism is in broad agreement with previous proposals for the reaction of Co(II)-substituted BcII with a range of β-lactam substrates,34,36 but differs substantially from the linear scheme that has been proposed to account for the reaction of the B2 MBL ImiS with the carbapenem substrate imipenem.37 We conclude that, at least for reaction of di-Co(II) enzyme with meropenem, SPM-1 should be considered as mechanistically close to B1, rather than B2, MBLs.
Conclusions
The combined results reveal that, in terms of its Zn(II) usage and catalytic mechanism, SPM-1 is best categorized as a B1 MBL. However, in structural terms, and in particular with respect to its flexible α3 region, SPM-1 has features typical of a B2 MBL. Interestingly, while SPM-1 is structurally unique amongst known MBLs, the α3 region that it shares with B2 MBLs has recently been observed in other members of the MBL superfamily, including an unusual type II glyoxylase from Salmonella enterica38 and the MBL fold protein TTHA163 from the marine thermophile Thermus thermophilus HB8.39 The growing prevalence of SPM-1 in carbapenem-resistant P. aeruginosa isolates in South America makes it one of the most clinically important MBLs, yet its unusual structural properties distinguish it from other B1 enzymes of equivalent medical relevance. In consequence, SPM-1 presents a unique challenge to inhibitor discovery programmes, and its inclusion in such studies has the potential to significantly complicate the search for inhibitors of all clinically significant enzymes. The results presented here go some way to identifying interaction sites, including a ‘hydrophobic wall’ at the active site that may be exploited in SPM-1 inhibitor discovery.Acknowledgements
We thank the Medical Research Council (MRC)/Canadian Grant G1100135 for support of J.B. and C.J.S. and Cancer Research UK (CRUK) for support of S.S.v.B and C.J.S. We thank the Dulverton Trust (A.M.R.), the Engineering and Physical Sciences Research Council (EPSRC) (W.B.S.), a Clarendon–St Hugh's College-Louey Scholarship (H.T.), a Royal Society Dorothy Hodgkin Fellowship (E.F.) and a Royal Society University Research Fellow (J.L.P.B.).References
- W. Lee, M. A. McDonough, L. Kotra, Z. H. Li, N. R. Silvaggi, Y. Takeda, J. A. Kelly and S. Mobashery, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1427–1431 CrossRef CAS PubMed.
- T. R. Walsh, M. A. Toleman, L. Poirel and P. Nordmann, Clin. Microbiol. Rev., 2005, 18, 306–325 CrossRef CAS PubMed.
- K. Bush and G. A. Jacoby, Antimicrob. Agents Chemother., 2010, 54, 969–976 CrossRef CAS PubMed.
- L. N. Andrade, N. Woodford and A. L. Darini, Int. J. Antimicrob. Agents, 2014, 43, 196–197 CrossRef CAS PubMed.
- A. E. Salabi, M. A. Toleman, J. Weeks, T. Bruderer, R. Frei and T. R. Walsh, Antimicrob. Agents Chemother., 2010, 54, 582 CrossRef PubMed.
- G. Cornaglia, H. Giamarellou and G. M. Rossolini, Lancet Infect. Dis., 2011, 11, 381–393 CrossRef CAS.
- M. Galleni, J. Lamotte-Brasseur, G. M. Rossolini, J. Spencer, O. Dideberg and J. M. Frere, Antimicrob. Agents Chemother., 2001, 45, 660–663 CrossRef CAS PubMed.
- A. I. Karsisiotis, C. F. Damblon and G. C. K. Roberts, Metallomics, 2014, 6, 1881–1197 RSC.
- M. A. Toleman, A. M. Simm, T. A. Murphy, A. C. Gales, D. J. Biedenbach, R. N. Jones and T. R. Walsh, J. Antimicrob. Chemother., 2002, 50, 673–679 CrossRef CAS PubMed.
- M. Polotto, T. Casella, M. G. de Lucca Oliveira, F. G. Rubio, M. L. Nogueira, M. T. de Almeida and M. C. Nogueira, BMC Infect. Dis., 2012, 12, 176 CrossRef PubMed.
- M. A. Toleman and T. R. Walsh, FEMS Microbiol. Rev., 2011, 35, 912–935 CrossRef CAS PubMed.
- A. P. Zavascki, P. B. Gaspareto, A. F. Martins, A. L. Goncalves and A. L. Barth, J. Antimicrob. Chemother., 2005, 56, 1148–1151 CrossRef CAS PubMed.
- T. A. Murphy, A. M. Simm, M. A. Toleman, R. N. Jones and T. R. Walsh, Antimicrob. Agents Chemother., 2003, 47, 582–587 CrossRef CAS.
- T. A. Murphy, L. E. Catto, S. E. Halford, A. T. Hadfield, W. Minor, T. R. Walsh and J. Spencer, J. Mol. Biol., 2006, 357, 890–903 CrossRef CAS PubMed.
- C. Moali, C. Anne, J. Lamotte-Brasseur, S. Groslambert, B. Devreese, J. Van Beeumen, M. Galleni and J.-M. Frère, Chem. Biol., 2003, 10, 319–329 CrossRef CAS.
- H. Zhang and Q. Hao, FASEB J., 2011, 25, 2574–2582 CrossRef CAS PubMed.
- G. Garau, C. Bebrone, C. Anne, M. Galleni, J. M. Frere and O. Dideberg, J. Mol. Biol., 2005, 345, 785–795 CrossRef CAS PubMed.
- C. Bebrone, C. Anne, F. Kerff, G. Garau, K. De Vriendt, R. Lantin, B. Devreese, J. Van Beeumen, O. Dideberg, J. M. Frere and M. Galleni, Biochem. J., 2008, 414, 151–159 CrossRef CAS PubMed.
- N. Sharma, Z. Hu, M. W. Crowder and B. Bennett, J. Am. Chem. Soc., 2008, 130, 8215–8222 CrossRef CAS PubMed.
- J. H. Toney, G. G. Hammond, P. M. Fitzgerald, N. Sharma, J. M. Balkovec, G. P. Rouen, S. H. Olson, M. L. Hammond, M. L. Greenlee and Y. D. Gao, J. Biol. Chem., 2001, 276, 31913–31918 CrossRef CAS PubMed.
- C. Bebrone, Biochem. Pharmacol., 2007, 74, 1686–1701 CrossRef CAS PubMed.
- A. M. Davies, R. M. Rasia, A. J. Vila, B. J. Sutton and S. M. Fabiane, Biochemistry, 2005, 44, 4841–4849 CrossRef CAS PubMed.
- J. L. Benesch and B. T. Ruotolo, Curr. Opin. Struct. Biol., 2011, 21, 641–649 CrossRef CAS PubMed.
- J. L. Benesch and C. V. Robinson, Curr. Opin. Struct. Biol., 2006, 16, 245–251 CrossRef CAS PubMed.
- A. M. Rydzik, J. Brem, S. S. van Berkel, I. Pfeffer, A. Makena, T. D. W. Claridge and C. J. Schofield, Angew. Chem., Int. Ed., 2014, 53, 3129–3133 CrossRef CAS PubMed.
- A. D. Bain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef CAS PubMed.
- A. D. Bain, Ann. Rep. NMR Spectrosc., 2008, 63, 23–48 CrossRef CAS.
- A. D. Bain and J. A. Cramer, J. Magn. Reson., Ser. A, 1993, 103, 217–222 CrossRef CAS.
- U. Heinz, R. Bauer, S. Wommer, W. Meyer-Klaucke, C. Papamichaels, J. Bateson and H. W. Adolph, J. Biol. Chem., 2003, 278, 20659–20666 CrossRef CAS PubMed.
- B. M. Lienard, G. Garau, L. Horsfall, A. I. Karsisiotis, C. Damblon, P. Lassaux, C. Papamicael, G. C. Roberts, M. Galleni, O. Dideberg, J. M. Frere and C. J. Schofield, Org. Biomol. Chem., 2008, 6, 2282–2294 CAS.
- A. Makena, S. S. van Berkel, C. Lejeune, R. J. Owens, A. Verma, R. Salimraj, J. Spencer, J. Brem and C. J. Schofield, ChemMedChem, 2013, 8, 1923–1929 CrossRef CAS PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. G. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- E. G. Orellano, J. E. Girardini, J. A. Cricco, E. A. Ceccarelli and A. J. Vila, Biochemistry, 1998, 37, 10173–10180 CrossRef CAS PubMed.
- M. F. Tioni, L. I. Llarrull, A. A. Poeylaut-Palena, M. A. Marti, M. Saggu, G. R. Periyannan, E. G. Mata, B. Bennett, D. H. Murgida and A. J. Vila, J. Am. Chem. Soc., 2008, 130, 15852–15863 CrossRef CAS PubMed.
- P. A. Crawford, K. W. Yang, N. Sharma, B. Bennett and M. W. Crowder, Biochemistry, 2005, 44, 5168–5176 CrossRef CAS PubMed.
- R. Bicknell, E. L. Emanuel, J. Gagnon and S. G. Waley, Biochem. J., 1985, 229, 791–797 CAS.
- N. P. Sharma, C. Hajdin, S. Chandrasekar, B. Bennett, K. W. Yang and M. W. Crowder, Biochemistry, 2006, 45, 10729–10738 CrossRef CAS PubMed.
- A. L. Stamp, P. Owen, K. El Omari, C. E. Nichols, M. Lockyer, H. K. Lamb, I. G. Charles, A. R. Hawkins and D. K. Stammers, Protein Sci., 2010, 19, 1897–1905 CrossRef CAS PubMed.
- A. Yamamura, A. Okada, Y. Kameda, J. Ohtsuka, N. Nakagawa, A. Ebihara, K. Nagata and M. Tanokura, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2009, 65, 455–459 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for protein expression and purification, 19F-labelling, crystallisation, data collection, and structure determination, table of crystallographic data, table of crystallographic parameters and refinement statistics, figures showing binding mode and distances, procedures for mass spectrometry measurements, differential scanning fluorimetry measurements, stopped-flow measurements and other kinetics measurements. See DOI: 10.1039/c4sc01752h |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |