
Comment on “Rabbit-ears hybrids, VSEPR sterics, and other orbital anachronisms”. A reply to a criticism
Philippe C.
Hiberty
*a,
David
Danovich
b and
Sason
Shaik
*b
aLaboratoire de Chimie Physique, CNRS UMR 8000, Université de Paris-Sud, 91405 Orsay Cédex, France. E-mail: philippe.hiberty@u-psud.fr; Fax: +33-0169154447; Tel: +33-0169156175
bThe Institute of Chemistry and the Lise Meitner-Minerva Center for Computational Quantum Chemistry, The Hebrew University, Jerusalem, 91904, Israel. E-mail: sason.shaik@gmail.com; Fax: +972-26584680; Tel: +972-26585909
First published on 13th March 2015
In a recent paper published in this Journal, and entitled “Rabbit-ears, VSEPR sterics, and other orbital anachronisms” (Clauss et al., 2014; hereafter referred to in brief as CNAMLW), Weinhold et al. criticize the concept of equivalent localized lone pairs, so-called the “rabbit-ears”, and in so doing they also specifically attack our own usage of these hybrid orbitals for H2O in chapter 5.7 of our book (Shaik and Hiberty, 2008). CNAMLW call the equivalent hybrid orbitals as “orbital anachronisms” and propose new hybrid orbitals based on their natural bond orbital (NBO) approach as being the sole legitimate localized Lewis-like representations of the water lone pairs. The authors further recommend changing the teaching curriculum, basing it on the new NBO hybrids, and discarding the “anachronistic” ones. Their critique of our book chapter, chosen as representative of the usage of equivalent lone pairs representation, is summarized as follows: (1) equivalent lone pairs obtained by transformations of the σ–π ones are claimed by CNAMLW to be far from sp3, being exceedingly p-rich; (2) equivalent lone pairs are claimed to be invalid as being not-symmetry-adapted; (3) the mathematical equivalence of equal-energy lone pairs to nonequivalent σ- and π-lone pairs of water is claimed to be untrue except at very low levels of the theory (actually Hückel-like, from Weinhold and Landis, 2012). The present comment summarizes our basic disagreements with the CNAMLW paper. It shows that the equivalent lone-pair picture (“rabbit-ears”) is an equivalent and a valid alternative to the nonequivalent one; neither is superior to the other or anachronistic with respect to the other.We read the CNAMLW paper with great surprise, since the commonly used “rabbit-ear” localized orbitals of H2O are derived by a transformation of the delocalized molecular orbitals (so-called canonical) in a manner that does not change the molecular wavefunction, nor its total energy nor any other observable properties. Thus, “rabbit-ear” lone pair orbitals and more generally localized orbitals constitute a perfectly valid set of orbitals, which are widely employed as standardly available by localization methods like Boys localization, Edmiston–Ruedenberg localization, etc., which are implemented in most of the widely-used quantum chemistry softwares. Anyone can get these hybrids by pressing a button! As such, saying, as CNAMLW do, that the equivalent lone pair representation of H2O is wrong or anachronistic is a misleading error that necessitates this comment. Impressing this wrong view as CNAMLW do as the sole legitimate opinion for teaching, this view becomes also misleading to chemistry teachers. We therefore show here that this opinion is based on erroneous calculations and logical inconsistencies.
To provide the reader with the necessary background, we first summarize briefly the frequently used sets of orbitals that can meaningfully describe a molecular electronic state, and subsequently, we examine the CNAMLW's critiques in detail.
Canonical MOs, Boys-localized MOs, natural bond orbitals. A reminder
Any standard molecular orbital calculation (so-called Hartree–Fock calculation) provides a set of molecular orbitals, called “canonical molecular orbitals” (CMOs), each of which has the physical meaning of being the mono-electronic wave function of a single electron in the average field of other electrons and nuclei. As such, CMOs are uniquely defined, and are also symmetry-adapted, i.e., they behave as irreducible representations of the symmetry group of the molecular species. These are the MOs that directly arise from MO calculations at any level, from Hückel to accurate calculations, and they have the nice property that their energies nearly correspond to the various molecular ionization potentials (IPs) as measured by photoelectron spectroscopy. Now, as these CMOs are generally delocalized over the whole molecule, they have little relevance to the Lewis representation of the molecule. Since chemists like to view molecules with individual bonds, it is necessary to generate also localized molecular orbitals (LMOs) that represent the molecule as a set of local two-electron bonds and lone pairs. To achieve this, one uses the property known from the early days of MO theory that the Hartree–Fock many-electron wave function is invariant to certain transformations among the occupied orbitals (Fock, 1930). These are called unitary transformations, which are nothing else than simple replacements of pairs of MOs by their sums and differences. One popular method to get rigorously defined LMOs is to transform the orbital set to localized orbitals, as in the Foster–Boys method (Boys, 1968). As a result, in the H2O case, the oxygen atom displays four near-sp3 hybrid atomic orbitals that form two equivalent lone pairs (Fig. 1a) and two equivalent O–H bonding LMOs (Fig. 1b) pointing to nearly tetrahedral directions. The resulting Foster–Boys localized lone-pair hybrids and LMOs displayed look indeed like manifestations of the Lewis-like structures, which are embedded in the CMOs of the Hartree–Fock wavefunction, and which are brought to light by the transformation. One good reason why this representation of water is popular is that equivalent lone pairs exactly predict the directions of hydrogen bonds that this molecule can have with others in space (see below).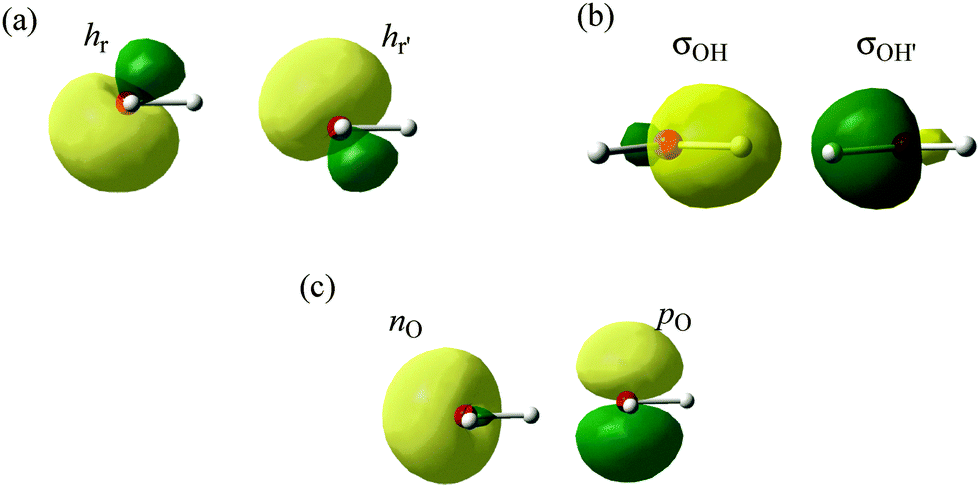 | ||
| Fig. 1 (a) The Boys-localized lone-pair hybrids (the rabbit-ears) without imposing σ–π separation. (b) The two O–H localized orbitals obtained either from Boys LMOs or from Weinhold NBOs. (c) The natural bond orbital (NBO) lone-pairs. | ||
Another way of getting localized orbitals is by calculating natural bond orbitals (NBOs), which, like CMOs, are also uniquely defined (for details, see the Appendix). Thus, NBOs for the O–H bonds of H2O are very similar to the corresponding LMOs σOH and σOH′ (Fig. 1b), and take the form of near-sp3 hybrids on oxygen, forming local bonds with hydrogens. On the other hand, the lone pairs NBOs keep the σ–π separation of the CMOs and yield a pure 2pO orbital perpendicular to the H2O plane, and a coplanar nO s-rich hybrid orbital (Fig. 1c). Finally, the σ–π lone-pairs are not specific to the NBO method, and can be obtained from the Boys-localization procedure by imposing σ–π separation as an option in the localization process, in which case NBOs and Boys localized lone-pairs are quite similar. In fact, as we repeatedly say in this comment, the sp3 lone pairs and the nonequivalent NBO lone pairs are related by a special transformation, called by mathematicians “unitary”, which in this case is particularly simple: if one just takes the sum and difference of the lone pairs in one set, one gets the lone pairs of the other set, as shown in Scheme 1. It is a two-way street! Both pictures are entirely equivalent and valid (for more details, a mathematical demonstration of the equivalence between the sp3-LMO and σ–π NBO lone pair representations at the ab initio level is shown in the Appendix below).
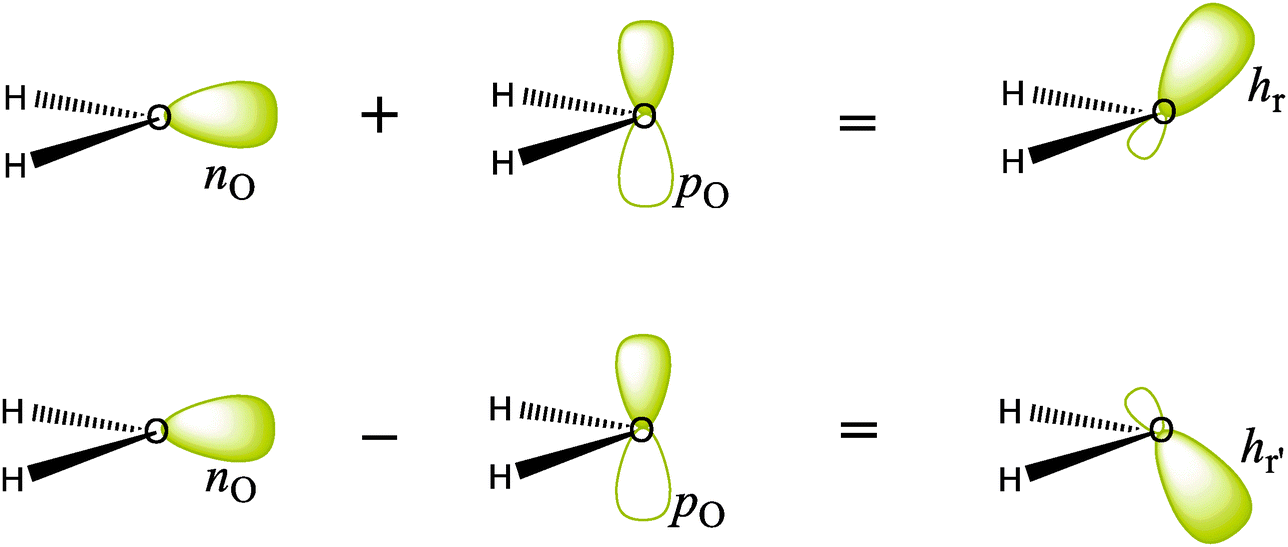 | ||
| Scheme 1 Transformation of the nonequivalent NBO σ–π lone pairs of H2O to get the equivalent sp3 lone pairs, by simple sum and difference. The back-transformation can be done in the same manner. | ||
Let us now examine in detail the critiques contained in the CNAMLW paper.
The allegedly excessive p-character of the LMOs lone pairs
Appendix 1 of the CNAMLW paper is a direct critique against Section 5.7 of the Shaik-Hiberty book (Shaik and Hiberty, 2008), which deals with the localized lone pairs (“rabbit-ears”) of H2O. In what seems to be an attempt at creating the impression that these localized lone pairs are very far from the expected sp3 form, the CNAMLW authors carry out the classical unitary mix of the NBO σ hybrid and pure p lone pairs (nO and pO, respectively, Fig. 1c) to form to the equivalent hybrids hr and hr′. Starting from the hypothesis that the nO hybrid has an sp2 hybridization, they apply the principle of conservation of p-character. Since the starting orbitals are supposed to have p-characters of 67% (for sp2) and 100% (for p), respectively, the respective p-characters of the equivalent hybrids, after unitary transformation, sum up to 167%. As such, CNAMLW deduce that each of the hr and hr′ localized lone pairs, involves 83% p-character, which is excessive and would correspond to hybrids of the sp4.9 type. All this argument rests on the assumption that the starting nO lone pair is effectively sp2. However this arbitrary assumption is erroneous. Indeed, the hybridization of the nO lone pair was accurately calculated by the NBO method and reported to be not sp2 but sp0.87 (46.5% p-character), in a recent book chapter (Landis and Weinhold, 2014). Moreover, this estimation was published by the same senior authors as those of the CNAMLW paper!This accurate result is in strong contradiction with the unfounded sp2 hypothesis of the CNAMLW paper. Let us now repeat the above qualitative transformation of the pO and nO lone pairs, using the actual hybridization parameter (0.87) provided by NBO for the latter. Conserving the total p-character of (0.87/1.87 = 0.465) for nO and 1.0 for pO, the equivalent lone pairs resulting from the transformation have 73.3% p-character each, which corresponds to hybrids of the type sp2.75, close to sp3 like the σOH hybrids, albeit slightly less p-rich, in agreement with Bent's rule (Bent, 1961). Further, we can use Coulson's directionality theorem (eqn (A4) in the CNAMLW paper) to show that the axes of the equivalent lone pairs both display an angle of 56° above or below the H2O plane, in excellent agreement with the experimental directions (57°) of the two equivalent hydrogen-bonds that can be formed from the oxygen to neighboring H atoms (Scheme 2).
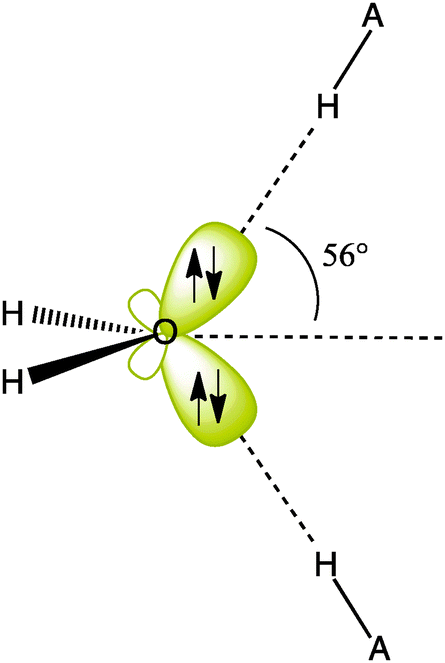 | ||
| Scheme 2 The LMO near-sp3 representation of the H2O lone pairs which is obtained by transforming (taking sum and difference) the nO and pO NBO lone pairs. Note that the directions of the lone pairs in the sp3 representation match the directions of hydrogen bonds between the oxygen atom of H2O and neighboring A–H molecules. | ||
Do localized lone pairs need be symmetry-adapted?
CNAMLW claim that localized lone pair orbitals, and in particular NBOs, must exhibit strict σ–π separation, as irreducible representations of C2v symmetry. However, this requirement does not apply to the orbitals that are used to describe the total wave function. Thus, both orbital pictures are equivalent and both are perfectly valid, and their choice of usage can be done according to the problem at hand. Incidentally, the CNAMLW argument about symmetry is particularly puzzling, as some of the NBOs that they advocate, namely the OH-bonding NBOs in Fig. 1b, are not irreducible representations of the C2v group!Are equivalent localized lone pairs in conflict with spectroscopic properties?
In their Appendix 1, CNAMLW also claim that the energy degeneracy of the equivalent LMO lone pairs is in conflict with spectroscopic properties, a hint to the fact that CMOs energies correspond to the ionization potentials (IPs) of H2O, while equivalent lone pair energies do not. Actually, the same critique of CNAMLW could have been directed at the NBO nO lone pair, since this orbital is not equivalent to any of the CMOs and therefore has an energy that does not correspond to any H2O IP. In fact, LMOs are never required to lead to IPs. The IPs are energy differences between the states of the molecule and its cation, and the wave functions that represent these states can use CMOs or LMOs (Shaik and Hiberty, 2008; Hiberty et al., 2012).Is the validity of the unitary transformation restricted to low theoretical levels?
Another critique against equivalent lone pairs argues as follows in the CNAMLW paper: “the supposed ‘equivalence’ of (n,p) vs. (hr,hr′) lone pairs rests on approximations that are unacceptable by current standards of accuracy”. This critique is rather surprising, given the universally recognized invariance of an ab initio many-electron wave function (Hartree–Fock) under unitary transformation of its orbitals. In fact, this critique is based on the fact that the Fock matrix interaction element between equivalent sp3 lone pairs is non-zero (and would be zero at, e.g. the Hückel level). However, the nullity of Fock matrix interaction elements between orbitals is a property that is fully satisfied only by CMOs in general (with some exceptions due to symmetry reasons) and is not at all required for a wave function to be valid. Would such a property be required, all localization methods, including the NBO method, would be invalid! Indeed, ab initio calculations show that the NBO orbitals σOH and σOH′ do have a non-zero Fock matrix interaction element, while being orthogonal to each others, just like the LMO lone pairs. Yet, the NBOs are still considered perfectly valid by CNAMLW who by the same criteria deny the validity of the localized lone pairs.The general validity of unitary transformations
CNAMLW question the validity of the concept of unitary transforms of MOs, using as an example hypothetical mixtures of core and valence orbitals, and argue that these mixtures “provide no real justification for claiming that core and valence orbitals are somehow equivalent”. However, unitary transforming σ–π lone pairs to equivalent sp3 lone pairs is not comparable to mixing of core to valence orbitals: in the first case we transform lone pairs to lone pairs, while in the second case we would transform core and valence orbitals to undefined and physically dubious mixtures which would be neither of the core or of the valence type. This is a bit of a strawman argument, since no chemist would think to create such core–valence hybrids.Conclusion
It follows from what precedes that one must give up the belief that there exists a unique set of supposedly “real”, or “best”, orbitals for a given molecule, e.g. the lone pairs of H2O. In fact, different sets of orbitals may describe the same reality (embedded in the many-electron wave function of any molecule). This view is held by all leading authorities in the field (Fock, 1930; Edmiston and Ruedenberg, 1963; Honneger and Heilbronner, 1991; Boys, 1968; Truhlar, 2012, and many others). Thus, the σ–π representation of lone pairs for H2O, and more generally of an O–R substituent, is evidently the best choice if this substituent is attached to a polyenic system and interacts with the π orbitals of the latter (as e.g., in furan). On the other hand, for an H2O molecule having both its lone pairs involved in hydrogen bonds with two external identical molecules (Scheme 2), the equivalent near-sp3 lone pair representation is the obvious choice. Thus, each set of lone pair orbitals has its own preferred domain of application, but they all lead to the same molecular properties and to the same unique electron density distribution in 3D space. Interestingly, such a density map exhibits clear “rabbit-ear” shapes for the lone pairs of H2O (see, e.g., http://people.nas.nasa.gov/%E2%88%BCcreon/papers/mgms96/), as revealed by the quantum theory of Atoms-in-Molecules (Bader, 1994). The CNAMLW strong recommendation to use only σ–π NBO lone pairs while discarding the sp3 LMO ones lacks therefore much if any scientific basis.Appendix
1. Unitary transformation of σ–π NBO lone pairs to near-sp3 LMO lone pairs for H2O
The equivalence between the many-electron wave functions constructed with either σ–π NBO lone pairs or near-sp3 LMO lone pairs for H2O is shown in the following development in which only the lone pairs are explicitly considered.Let us call n and p, respectively, the σ and π inequivalent lone pairs provided by the NBO method or by the Boys-localization method with σ–π separation (Fig. 1c). The equivalent LMO lone pairs, hr and hr′, are obtained as the sum and difference of n and p:
| hr = 2−1/2(n + p) | (1a) |
| hr′ = 2−1/2(n − p) | (1b) |
ΨLMO = |hr![[h with combining macron]](https://www.rsc.org/images/entities/char_0068_0304.gif) rhr′r′| = ¼(|(n + p)( rhr′r′| = ¼(|(n + p)(![[n with combining macron]](https://www.rsc.org/images/entities/char_006e_0304.gif) + + ![[p with combining macron]](https://www.rsc.org/images/entities/char_0070_0304.gif) )(n − p)( − )|) = ¼(|np| − |np| − |pn| + |pn|) = |np| )(n − p)( − )|) = ¼(|np| − |np| − |pn| + |pn|) = |np| | (2) |
2. Natural bonding orbitals
Natural bonding orbitals (NBOs) are uniquely defined via diagonalization of the one-electron reduced density matrix that is provided by any MO- or DFT-based computational methods, while restricting the MOs to atomic regions or diatomic bonding regions (Weinhold and Landis, 2005). As such, they are different from the CMOs, which are also uniquely defined, but by another criterion, as eigenfunctions of the Fock operator. Unlike LMOs, NBOs can be obtained either from simple Hartree–Fock wave functions or from more complex ones, or from DFT calculations. This however does not make much difference in the frequent case of molecular ground states, which are not significantly multi-reference (e.g., H2O).References
- Bader R., (1994), Atoms in Molecules: A Quantum Theory, USA: Oxford University Press.
- Bent H. A., (1961), An appraisal of valence-bond structures and hybridization in compounds of the first-row elements, Chem. Rev., 61, 275–311.
- Boys S. F., (1968), in Löwdin P.-O. (ed.), Quantum Theory of Atoms, Molecules, and the Solid State, New York: Academic Press, p. 253.
- Clauss A. D., Nelsen S. F., Ayoub M., Moore J. W., Landis C. R. and Weinhold J. W., (2014), Rabbit-ears hybrids, VSEPR sterics, and other orbital anachronisms, Chem. Educ. Res. Pract., 15, 417–434.
- Edmiston C. and Ruedenberg K., (1963), J. Rev. Mod. Phys., 35, 457–465.
- Fock V., (1930), Approximation methods for solution of the quantum mechanical many-body problem (in German), Z. Physik, 61, 126–148.
- Hiberty P. C., Volatron F. and Shaik S., (2012), In Defense of the Hybrid Atomic Orbitals, J. Chem. Educ., 89, 575–577.
- Honneger E. and Heilbronner E., (1991), in Maksic Z. B. (ed.), Theoretical Models of Chemical Bonding, Berlin-Heidelberg: Springer-Verlag, vol. 3, pp. 100–151.
- Landis C. R., Weinhold F., (2014), in Frenking G. and Shaik S. (ed.), The Chemical Bond, Weinheim, Germany: Wiley-VCH, p. 91.
- Shaik S. and Hiberty P. C., (2008), A Chemist's Guide to Valence Bond Theory, Hoboken, NJ: Wiley-Interscience.
- Truhlar D. G., (2012), Are Molecular Orbitals Delocalized? J. Chem. Educ., 89, 573–574.
- Weinhold F. and Landis C. R., (2005), Valency and Bonding, London: Cambridge U. Press.
- Weinhold F. and Landis C. R., (2012), Discovering Chemistry with Natural Bond Orbitals, Hoboken, NJ: Wiley.
| This journal is © The Royal Society of Chemistry 2015 |