
Metal-free catalytic cascade to chromones: direct coupling of salicylaldehydes and activated alkynes triggered by aryloxyl radicals†
Ping Wang‡
ab,
Zhongfeng Li‡a,
Shengli Caoa and
Honghua Rao *a
*a
aDepartment of Chemistry, Capital Normal University, Beijing 100048, P. R. China. E-mail: honghua.rao@gmail.com
bSchool of Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing 102617, P. R. China
First published on 3rd December 2015
Abstract
The first intermolecular addition reaction of aryloxyl radicals to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds was disclosed, which can afford biologically important chromone scaffolds via a PhNMe3I-catalyzed direct coupling of readily available salicylaldehydes and activated internal alkynes in only one step, and can tolerate a range of catalytically reactive functional groups.
C bonds was disclosed, which can afford biologically important chromone scaffolds via a PhNMe3I-catalyzed direct coupling of readily available salicylaldehydes and activated internal alkynes in only one step, and can tolerate a range of catalytically reactive functional groups.
Chromones are ubiquitous structural scaffolds occurring in numerous natural products, pharmaceutical agents, and drug candidates that manifest high levels of various important biological activities.1 For example, the 3-carboxylated chromone analogues and their carboxamide derivatives can act as potent and reversible monoamine oxidase-B inhibitors,2 while 3-carboxylated flavones are protein-tyrosine kinase inhibitors3 and antiinflammatories.4 Due to the significant biological activities of these privileged structures, a variety of strategies have been developed for the construction, functionalization, and diversification of chromone scaffolds. Conventionally, intramolecular cyclization of the previously synthesized intermediates, such as ortho-hydroxyaryl-1,3-dicarbonyl derivatives (or precursors) (Fig. 1a),5 2′-hydroxychalcones (Fig. 1b),6 2-aryloxymaleates (Fig. 1c),7 or vinyl ether intermediates of salicylic acid derivatives (Fig. 1d),8 can afford functionalized chromones efficiently under strong acidic or basic conditions.
 | ||
| Fig. 1 Chromones from various intermediates or starting materials via conventional or catalyzed cyclization strategies. | ||
To avoid multistep procedure for the construction of chromones, palladium catalysis (Fig. 1e)9 and [RhCl(cod)]2 (Fig. 1f)10 have been proposed, respectively. However, as trace metals in products for human consumption are heavily regulated by international bodies, the concerns associated with removing potentially toxic trace transition-metal catalysts from the end biologically active products always exist in drug discoveries.11 For this reason, organocatalyzed reactions using N-heterocyclic carbenes (NHCs) are developed for the green syntheses of chromones, although pre-functionalizations of hydroxyl groups on salicylaldehydes are still required.12 Thus, it remains desirable to develop the organocatalyzed reactions that can realize the chromone syntheses through direct couplings of readily available starting materials in an atom- and step-economical way.
As is well recognized, radical cascade reactions, usually initiated by simple organic molecules, can allow access to complex and important molecular skeletons in only few synthetic steps.13 During the last decades, intermolecular radical addition to CC π-system generating reactive vinyl radicals to perform intramolecular cascade cyclizations have been well established with carbon-,13b,14 sulphur-,13b,15 and oxygen-centered organic radicals (typically TEMPO and its analogues, acyloxyl radicals, or methoxyl radical).16 In this context, the synthetically attractive strategy may also enable a rapid obtention of chromones if a cascade cyclization could be initiated by the intermolecular addition of a specific oxygen-centered radical, namely the 2-formylaryloxyl radical, to CC bonds. We herein report a radical cascade initiated by the intermolecular addition of 2-formylaryloxyl radical to CC π-system for the first time, which can afford chromones in moderate to good yields via a PhNMe3I-catalyzed direct coupling of readily available salicylaldehydes and activated alkynes (Scheme 1).
 | ||
| Scheme 1 PhNMe3I-catalyzed intermolecular addition of 2-formylaryloxyl radical to alkynes. | ||
At the outset of our study, an intermolecular radical addition of phenol to dimethyl acetylenedicarboxylate (DMAD) was successfully realized under nBu4NI/TBHP organocatalytic system in our group. Following this initial discovery, salicylaldehyde 1a (0.10 mmol) and DMAD 2a (0.15 mmol) were chosen as the model substrates to test the possibility of chromone formation, with TBAI (10 mol%) as the catalyst, TBHP (2.5 equiv.) as the oxidant, and CH3CN (1.0 mL) as the solvent. As shown in Table 1, inspiringly, trace amount of chromone product 3a was detected when using TBHP (70 wt% in H2O) as the oxidant (Table 1, entry 1), and the preliminary experiments identified TBHP (5–6 M in decane) as the oxidant of choice under an air atmosphere (Table 1, entries 1–3). Notably, nearly no product 3a was detected in the absence of catalyst TBAI or oxidant TBHP (entries 4–5), thus suggesting that it is crucial for this transformation to use ammonium salt in combination with oxidant. And it is noteworthy that PhNMe3I displayed the best catalytic reactivity among a variety of quaternary ammonium salts, such as TBAI, THAI, TEAI, and BnNMe3I, affording the desired product 3a in 74% yield (entries 2, and 6–9). Besides, the reaction efficiency shows a strong dependency on solvent. For example, the use of other solvents, such as toluene, benzene, DCE, TCE, tBuOH, and EtOAc, proved to be far less effective than acetonitrile (entries 8, and 10–15). Other endeavours to improve the reaction efficiency were attempted as well, such as loading 5% or 20% of the catalyst (entries 16 and 17), changing the amount of TBHP (5–6 M in decane) (entry 18), or carrying out the reaction at 110 °C or 130 °C (entries 19 and 20), but none of them afforded superior result.
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | [Ox] (equiv.) | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.15 mmol), solvent (1.0 mL), under air for 24 h unless otherwise noted.b Yield determined by 1H NMR spectroscopy with mesitylene as the internal standard.c Under N2.d Dimethyl 2-(2-formylphenoxyl)-2-butenedioate detected as the major byproduct in 10% yield. [Ox] = oxidant, TBAI = tetrabutylammonium iodide, THAI = tetra-n-heptylammonium iodide, TEAI = tetraethylammonium iodide, TBHPaq = tert-butyl hydroperoxide (70 wt% in H2O), TBHPdec = tert-butyl hydroperoxide (5–6 M in decane), DCE = 1,2-dichloroethane, TCE = 1,1,2-trichloroethane. | |||||
| 1 | TBAI (10) | TBHPaq (2.5) | CH3CN | 120 | <5 |
| 2 | TBAI (10) | TBHPdec (2.5) | CH3CN | 120 | 43 |
| 3c | TBAI (10) | TBHPdec (2.5) | CH3CN | 120 | 23 |
| 4 | — | TBHPdec (2.5) | CH3CN | 120 | <5 |
| 5 | TBAI (10) | — | CH3CN | 120 | 0 |
| 6 | THAI (10) | TBHPdec (2.5) | CH3CN | 120 | 26 |
| 7 | TEAI (10) | TBHPdec (2.5) | CH3CN | 120 | 22 |
| 8 | PhNMe3I (10) | TBHPdec (2.5) | CH3CN | 120 | 74d |
| 9 | BnNMe3I (10) | TBHPdec (2.5) | CH3CN | 120 | 30 |
| 10 | PhNMe3I (10) | TBHPdec (2.5) | Toluene | 120 | 11 |
| 11 | PhNMe3I (10) | TBHPdec (2.5) | Benzene | 120 | 12 |
| 12 | PhNMe3I (10) | TBHPdec (2.5) | DCE | 120 | 12 |
| 13 | PhNMe3I (10) | TBHPdec (2.5) | TCE | 120 | 18 |
| 14 | PhNMe3I (10) | TBHPdec (2.5) | tBuOH | 120 | 23 |
| 15 | PhNMe3I (10) | TBHPdec (2.5) | EtOAc | 120 | 17 |
| 16 | PhNMe3I (5) | TBHPdec (2.5) | CH3CN | 120 | 59 |
| 17 | PhNMe3I (20) | TBHPdec (2.5) | CH3CN | 120 | 45 |
| 18 | PhNMe3I (10) | TBHPdec (2.0) | CH3CN | 120 | 54 |
| 19 | PhNMe3I (20) | TBHPdec (2.5) | CH3CN | 110 | 63 |
| 20 | PhNMe3I (20) | TBHPdec (2.5) | CH3CN | 130 | 59 |

With the optimized reaction conditions in hand, we probed the substrate scope at 120 °C for 24 h under an air atmosphere by using PhNMe3I (10 mol%) as the catalyst, TBHP (5–6 M in decane, 2.5 equiv.) as the oxidant, and CH3CN as the solvent. As summarized in Table 2, this PhNMe3I-catalyzed cascade reaction was successfully performed with a variety of salicylaldehydes and DMAD, affording useful chromones containing an ester group at C3 position2 in moderated to good yields. Apparently, the reaction proceeded well with electron-donating groups at salicylaldehydes, such as methyl (3b–d), methoxy (3e–f) and benzyloxy (3g) groups. Electron-withdrawing groups at salicylaldehydes were well tolerated for this cascade reaction as well, such as chloro (3h) and bromo (3i) groups, albeit in slightly lower yields of the desired chromone products. These useful electrophilic halide groups promise further functionalizations of the products, thus facilitating the diversification and post-modification of chromone scaffolds. It is worth noting that steric hindrance effect is observed in this radical cascade reaction, for example, 3-methyl group at salicylaldehyde gave 63% of the desired product 3b, while 73% of 3c with 4-methyl and 75% of 3d with 5-methyl, respectively. To expand the substrate scope, other internal alkynes were also tested, among which the symmetrical alkynes, such as diethyl acetylenedicarboxylate (3j–l) and dibenzoylacetylene (3m), could successfully provide the desired chromone scaffolds in good yields. Moreover, unsymmetrical alkyne substrates that would exhibit specific selectivity challenges during intermolecular radical additions were explored under the optimized conditions as well. Inspiringly, all the examined methyl arylpropiolates (3n–r) underwent the organo-mediated cascades smoothly and selectively to yield 3-carboxylated flavones3,4 as the sole products,17 with the catalytically reactive substituents (such as cyano and halide) intact during the process. Unfortunately, the cascade reaction did not occur when using less electron-deficient alkynes (such as 1,2-diphenylethyne, methyl propiolate, methyl 2-butynoate, 1-phenyl-1-propyne) or salicylaldehydes with strong electron-withdrawing groups (such as nitro and cyano groups)18 as the substrates. Therefore, for successful formation of chromone derivatives, more electron-deficient alkynes and electron-efficient salicylaldehydes are required.


To demonstrate the practicability of the developed strategy, we carried out a gram-scale synthesis of product 3a. The target product was isolated in 68% yield without any significant decrease in efficiency (versus 71% for the reaction on a 0.2 mmol scale for 3a). Besides, 3-carboxylated flavones can undergo efficient decarboxylative process to the desired biologically active flavones. For instance, reaction of product 3n with hydrobromic acid (48% aqueous solution) gave 92% of 7-hydroxyflavone 4 (Scheme 2).
 | ||
| Scheme 2 Decarboxylation of 3-carboxylated flavones: 3n (0.2 mmol), HBr (48% aqueous solution, 3 mL), 110–120 °C, under N2, 24 h. | ||
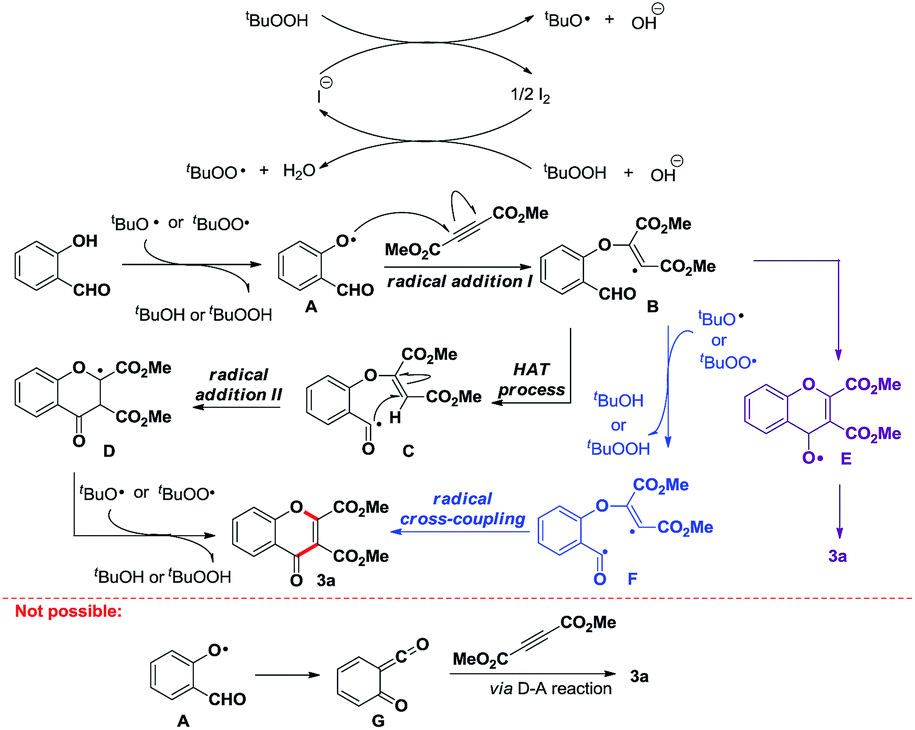
When investigating the mechanism for this process, a reaction of salicylaldehyde with DMAD was first carried out in the presence of K2CO3 (a popular base for Michael addition reaction) in CH3CN, and it gave only 10% of product 6, with vinyl ether 5a/5b as the major products, implying that a nucleophilic addition of vinyl anion (obtained via a Michael addition of phenolate to alkyne) to aldehyde group to form 4H-chromen-2,3-dicarboxylate (prone to be oxidized to chromone) is not a major pathway for the formation of chromones (Scheme 3-i). On the other hand, as it has been disclosed by our group19 and others,20 when using quaternary ammonium salts as the catalysts, tBuO˙ or tBuOO˙ can be generated efficiently from TBHP, and can abstract a hydrogen to initiate radical reactions subsequently. Therefore, a radical process is an alternative consideration, and the radical trapping experiment was then carried out by introducing TEMPO or BHT (2,6-di-tert-butyl-4-methylphenol) into the standard reaction conditions. Indeed, none of the product 3a, vinyl ether 5a/5b, or direct hydroacylation product of CC bond was observed (Scheme 3-ii), so that it may lead to two preliminary conclusions: (1) the reaction most probably undergoes either 2-formylaryloxyl or 2-hydroxybenzoyl radical initiated cascade cyclizations to chromones; (2) no detection of vinyl ether byproducts indicates that a polar addition of phenolate to alkynes might be impossible under our standard conditions.
 | ||
| Scheme 3 Control experiments for mechanism studies. | ||
In order to explore that whether this cyclization is initiated by the intermolecular addition of 2-hydroxybenzoyl radical to CC bond, reaction of o-anisaldehyde with DMAD was carried out under the standard conditions, however, no hydroacylation product 7 was detected (Scheme 3-iii), thus suggesting that this cyclization is probably not initiated by 2-hydroxybenzoyl radical. Alternatively, 2-formylaryloxyl radicals can be generated upon reactions of peroxyl or alkyloxyl radicals with corresponding phenols.18 As this aryloxyl radical might undergo a hydrogen-abstraction process to give 6-oxomethylidenecyclohexa-2,4-dienone analogues (structure G in Scheme 4), which can facilitate a facile Diels–Alder reaction with DMAD, a reaction of salicylaldehyde with dimethyl maleate was thereby carried out. However, none of the cyclization product 8 or 3a was detected (Scheme 3-iv), indicating that a Diels–Alder cyclization to chromones is neither possible in our reaction. As such, we can argue that this PhNMe3I-catalyzed cyclization reaction is initiated by an intermolecular addition of aryloxyl radical to CC bond, affording the reactive vinyl radical, which can undergo further cyclization to the end product 3 or give vinyl ether byproducts.
 | ||
| Scheme 4 Plausible mechanism for PhNMe3I-catalyzed intermolecular addition of aryloxyl radicals to CC bond. | ||
Finally, when vinyl ether 5a or 5b was directly subjected to the standard reaction conditions (an acyl radical generation system directly from aldehyde group),19,20 only 16% or 15% of the desired product 3a was detected, inferring that an intramolecular acyl radical addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is possible in the process (Scheme 3-v). But considering their much lower reaction efficiency, the acyl radical intermediate (structure C in Scheme 4) may be relatively difficult to be generated directly from vinyl ether 5a/5b in our catalytic system; instead it could form more efficiently via a direct intramolecular H-abstraction from aldehyde group by vinyl radical.
C bond is possible in the process (Scheme 3-v). But considering their much lower reaction efficiency, the acyl radical intermediate (structure C in Scheme 4) may be relatively difficult to be generated directly from vinyl ether 5a/5b in our catalytic system; instead it could form more efficiently via a direct intramolecular H-abstraction from aldehyde group by vinyl radical.
Based on above results and previous work, a tentative mechanism for this PhNMe3I-catalyzed radical cascade is depicted in Scheme 4. In the first step, tBuO˙ and tBuOO˙ radicals form catalytically.19,20 After abstracting a hydrogen from hydroxy group by these radicals,18 an intermolecular addition of the resulting aryloxyl radical A to CC bond occurs, thereby generating a reactive vinyl radical B, which prefers to abstract a hydrogen from aldehyde group intramolecularly to produce acyl radical C. The subsequent intramolecular addition of acyl radical C to CC bond forms a carbon-centered radical D, which finally produces product 3a via H-abstraction by tBuO˙ or tBuOO˙ radical. However, the pathway to form chromones via either intramolecular addition of radical B to aldehyde (forming radical E)21 or intramolecular coupling of acyl radical with vinyl radical (intermediate F in Scheme 4) cannot be ruled out at this stage.22
In summary, we have developed a PhNMe3I-catalyzed radical cascade to chromone scaffolds (particularly the useful 3-carboxylated chromones that cannot be synthesized conveniently with other reported strategies) via a single operation, which serves as the first example on intermolecular addition of aryloxyl radical to CC bond. The organocatalyzed reaction proceeds smoothly with readily available salicylaldehydes and activated internal alkynes, and can tolerate a range of catalytically reactive functional groups, thus may provide concise approaches to biologically important chromone skeletons in drug discoveries. Studies to clearly understand the mechanism and explore the potential applications of aryloxyl radicals are underway in our lab.
Acknowledgements
Financial support from The National Natural Science Foundation of China (21402128), Beijing Natural Science Foundation (2144045), Beijing Municipal Education Commission Foundation (KM201410028007), Scientific Research Base Development Program of the Beijing Municipal Commission of Education, and Capital Normal University are gratefully acknowledged.Notes and references
- (a) G. P. Ellis, in Chromenes, Chromanones, and Chromones: The Chemistry of Heterocyclic Compounds, ed. G. P. Ellis, John Wiley & Sons, Inc., New York, 1977, Vol. 31 Search PubMed; (b) A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960 CrossRef CAS PubMed , and references cited therein.
- For selected examples, see: (a) A. Gaspar, T. Silva, M. Yáñez, D. Vina, F. Orallo, F. Ortuso, E. Uriarte, S. Alcaro and F. Borges, J. Med. Chem., 2011, 54, 5165 CrossRef CAS PubMed; (b) A. Gaspar, J. Reis, A. Fonseca, N. Milhazes, D. Viña, E. Uriarte and F. Borges, Bioorg. Med. Chem. Lett., 2011, 21, 707 CrossRef CAS PubMed; (c) A. Gaspar, F. Teixeira, E. Uriarte, N. Milhazes, A. Melo, M. N. D. S. Cordeiro, F. Ortuso, S. Alcaro and F. Borges, ChemMedChem, 2011, 6, 628 CrossRef CAS PubMed; (d) A. Gaspar, J. Reis, S. Kachler, S. Paoletta, E. Uriarte, N. Klotz, K. S. Moro and F. Borges, Biochem. Pharmacol., 2012, 84, 21 CrossRef CAS PubMed; (e) L. J. Legoabe, A. Petzer and J. P. Petzer, Bioorg. Med. Chem. Lett., 2012, 22, 5480 CrossRef CAS PubMed.
- M. Cushman, D. Nagarathnam, D. L. Burg and R. L. Geahlen, J. Med. Chem., 1991, 34, 798 CrossRef CAS PubMed.
- R. Walenta, R. Mueller-Peddinghaus, I. Ban, M. Wurl and U. Preuschoff, Eur. Pat. Appl., 1988, EP290915 Search PubMed.
- For representative examples, see: (a) R. A. Appleton, J. R. Bantick, T. R. Chamberlain, D. N. Hardern, T. B. Lee and A. D. Pratt, J. Med. Chem., 1977, 20, 371 CrossRef CAS PubMed; (b) J. H. Looker, J. H. McMechan and J. W. Mader, J. Org. Chem., 1978, 43, 2344 CrossRef CAS; (c) C. A. Gray, P. T. Kaye and A. T. Nchinda, J. Nat. Prod., 2003, 66, 144 CrossRef PubMed; (d) D. A. Vasselin, A. D. Westwell, C. S. Matthews, T. D. Bradshaw and M. F. G. Stevens, J. Med. Chem., 2006, 49, 3973 CrossRef CAS PubMed; (e) S. J. Hosseinimehr, S. Emami, S. M. Taghdisi and S. Akhlaghpoor, Eur. J. Med. Chem., 2008, 43, 557 CrossRef CAS PubMed; (f) J. Zhao, Y. F. Zhao and H. Fu, Org. Lett., 2012, 14, 2710 CrossRef CAS PubMed; (g) J.-P. Lin and Y.-Q. Long, Chem. Commun., 2013, 49, 5313 RSC.
- For representative examples, see: (a) X. Huang, E. Tang, W. Xu and J. Cao, J. Comb. Chem., 2005, 7, 802 CrossRef CAS PubMed; (b) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070 CrossRef CAS PubMed; (c) C. A. Engelhart and C. C. Aldrich, J. Org. Chem., 2013, 78, 7470 CrossRef CAS PubMed.
- For representative examples, see: (a) D. Obrecht, Helv. Chim. Acta, 1989, 72, 447 CrossRef CAS; (b) L. Costantino, G. Rastelli, M. C. Gamberini, J. A. Vinson, P. Bose, A. Iannone, M. Staffieri, L. Antolini, A. Del Corso, U. Mura and A. Albasini, J. Med. Chem., 1999, 42, 1881 CrossRef CAS PubMed; (c) E. Fillion, A. M. Dumas, B. A. Kuropatwa, N. R. Malhotra and T. C. Sitler, J. Org. Chem., 2005, 71, 409 CrossRef PubMed.
- (a) F. Pochat and P. L′Haridon, Synth. Commun., 1998, 28, 957 CrossRef CAS; (b) P. Kumar and M. S. Bodas, Org. Lett., 2000, 2, 3821 CrossRef CAS; (c) J. Jung, J. Min and O.-S. Park, Synth. Commun., 2001, 31, 1837 CrossRef CAS; (d) G. Athanasellis, G. Melagraki, A. Afantitis, K. Makridima and O. Igglessi-Markopoulou, Arkivoc, 2006, 10, 28 Search PubMed.
- (a) C.-F. Lin, W.-D. Lu, I. W. Wang and M.-J. Wu, Synlett, 2003, 13, 2057 Search PubMed; (b) B. Liang, M. Huang, Z. You, Z. Xiong, K. Lu, R. Fathi, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 6097 CrossRef CAS; (c) E. Awuah and A. Capretta, Org. Lett., 2009, 11, 3210 CrossRef CAS; (d) Q. Yang and H. Alper, J. Org. Chem., 2010, 75, 948 CrossRef CAS; (e) M. Yoshida, Y. Fujino and T. Doi, Org. Lett., 2011, 13, 4526 CrossRef CAS PubMed; (f) X. Wang, G. Cheng and X. Cui, Chem. Commun., 2014, 50, 652 RSC.
- M. Shimizu, H. Tsurugi, T. Satoh and M. Miura, Chem.–Asian J., 2008, 3, 881 CrossRef CAS PubMed.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef.
- (a) S. Vedachalam, J. Zeng, B. K. Gorityala, M. Antonio and X.-W. Liu, Org. Lett., 2010, 12, 352 CrossRef CAS PubMed; (b) S. Vedachalam, Q.-L. Wong, B. Maji, J. Zeng and X.-W. Liu, Adv. Synth. Catal., 2011, 353, 219 CrossRef CAS; (c) L. Wen, H. Zhang, H. Lin, Q. Shen and L. Lu, J. Fluorine Chem., 2012, 133, 171 CrossRef CAS.
- (a) P. J. Parsons, C. S. Penkett and A. Shell, Chem. Rev., 1996, 96, 195 CrossRef CAS PubMed; (b) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed.
- (a) J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef CAS; (b) J. S. Xiang and P. L. Fuchs, Tetrahedron Lett., 1996, 37, 5269 CrossRef CAS; (c) J. S. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 119, 4123 CAS; (d) R. A. Doohan and N. W. A. Geraghty, Green Chem., 2005, 7, 91 Search PubMed; (e) Z.-Q. Liu, L. Sun, J.-G. Wang, J. Han, Y.-K. Zhao and B. Zhou, Org. Lett., 2009, 11, 1437 CrossRef CAS PubMed; (f) J.-Y. Luo, H.-L. Hua, Z.-S. Chen, Z.-Z. Zhou, Y.-F. Yang, P.-X. Zhou, Y.-T. He, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2014, 50, 1564 RSC; (g) J. Li, J. Zhang, H. Tan and D. Z. Wang, Org. Lett., 2015, 17, 2522 CrossRef CAS PubMed; (h) S. Manna and A. P. Antonchick, Org. Lett., 2015, 17, 4300 CrossRef CAS PubMed; (i) H. Wang and S. Yu, Org. Lett., 2015, 17, 4272 Search PubMed.
- (a) J. Zhou, G.-L. Zhang, J.-P. Zou and W. Zhang, Eur. J. Org. Chem., 2011, 3412 CrossRef CAS; (b) B. Gabriele, R. Mancuso, E. Lupinacci, L. Veltri, G. Salerno and C. Carfagna, J. Org. Chem., 2011, 76, 8277 CrossRef CAS PubMed; (c) Y. Lin, G. Lu, C. Cai and W. Yi, Org. Lett., 2015, 17, 3310 CrossRef CAS PubMed.
- For TEMPO and its analogues as radical initiators, see: (a) B. König, W. Pitsch, M. Klein, R. Vasold, M. Prall and P. R. Schreiner, J. Org. Chem., 2001, 66, 1742 CrossRef . For acyloxyl radical initiators, see: ; (b) U. Wille, J. Am. Chem. Soc., 2002, 124, 14 CrossRef CAS PubMed; (c) C. Jargstorff and U. Wille, Eur. J. Org. Chem., 2003, 3173 CrossRef CAS . For methoxyl radical initiator, see this referecne.
- 4-Methoxysalicyaldehyde 1n and phenylpropiolate were reacted under the standard reaction conditions. After the solvent was removed under vacuum directly from the reaction mixture without any other workup-procedures, the residue was treated with HBr (48% aqueous solution), and 7-hydroxyflavone 4 was then isolated in 42% yield with 63% recovery of phenylpropiolic acid, thus confirming the distinguished regioselectivity of this organocatalyzed reaction.
- Aryloxyl radicals with strong electron-withdrawing groups are difficult to be generated from the corresponding phenols, and are very unstable even under inert atmosphere, see: E. R. Altwicker, Chem. Rev., 1967, 67, 475 CrossRef CAS , this is also the reason why only very activated alkynes are suitable for this cyclization strategy.
- H. H. Rao, X. Ma, Q. Liu, Z. Li, S. Cao and C.-J. Li, Adv. Synth. Catal., 2013, 355, 2191 CrossRef CAS.
- (a) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (b) Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231 CrossRef CAS PubMed; (c) W.-P. Mai, H.-H. Wang, Z.-C. Li, J.-W. Yuan, Y.-M. Xiao, L.-R. Yang, P. Mao and L.-B. Qu, Chem. Commun., 2012, 48, 10117 RSC; (d) B. Tan, N. Toda and C. F. Barbas III, Angew. Chem., Int. Ed., 2012, 51, 12538 CrossRef CAS PubMed; (e) X. Li, M. Fang, P. Hu, G. Hong, Y. Tang and X. Xu, Adv. Synth. Catal., 2014, 356, 2103 CrossRef CAS; (f) Y. Lv, Y. Li, T. Xiong, Y. Lu, Q. Liu and Q. Zhang, Chem. Commun., 2014, 50, 2367 RSC.
- For examples on the addition of C-centered radical to carbonyl moieties to form O-centered radicals, see: (a) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 230 CrossRef CAS; (b) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 2674 CrossRef CAS.
- (a) For efficient intermolecular cross-coupling of radicals, see ref. 20b; (b) For efficient intramolecular cross-coupling of radicals, see: J.-K. Qiu, B. Jiang, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, J. Sun, P. Wei, S.-J. Tu and G. Li, J. Am. Chem. Soc., 2015, 137, 8928 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedure for synthesis, characterization data, and 1H, 13C, and 19F NMR spectra of compounds. See DOI: 10.1039/c5ra24634b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |