
Elastic, crosslinked poly(acrylic acid) filaments: nanocellulose reinforcement and graphene lubrication†
S. Spoljaric,
A. Salminen,
N. D. Luong and
J. Seppälä*
Polymer Technology Research Group, Department of Biotechnology and Chemical Technology, School of Chemical Technology, Aalto University, P. O Box 16100 Aalto, Finland. E-mail: jukka.seppala@aalto.fi
First published on 7th December 2015
Abstract
Hybrid monofilaments of poly(acrylic acid) (PAA) and 1,6-hexanediol diglycidyl ether (16DGE), compounded with nanofibrillated cellulose (NFC) and graphene, were thermally crosslinked and subsequently spun from aqueous solution. Crosslinking, in the form of ester linkage formation, between PAA and 16DGE was successfully achieved via thermal induction. The monofilaments were elastic and flexible in nature, displaying remarkable elongation and work-to-break values (up to nine times higher than pure PAA–16DGE filaments). This unique behaviour derives from a synergy between the fillers; namely the reinforcing ability of cellulose nanofibrils and the lubricating effect of graphene.
Introduction
Filaments and yarns derived from renewable, natural resources have received significant research and industrial attention within recent years. One major factor has been reducing the global dependence on cotton, due to the environmental and economic problems associated with its cultivation.1 Man-made filaments/yarns derived from regenerated cellulose (cellulose II) have been amongst the most numerous and promising candidates. Viscose has been in common use since the 1930s,2 while recent developments such as Lyocell3 and Ioncell-F4 have yielded impressive properties. However, the hazardous reagents utilised in their manufacture5 and brittle nature of most cellulosic materials has hindered the development of bio-based and renewable elastic and ductile filaments/yarns.A potential alternative involves the use of poly(acrylic acid)–diglycidyl ether systems to produce a gel-like material that can be readily drawn into filaments. These crosslinked systems have been utilised to form various gel structures,6–8 however no attention has been focused towards film or filament preparation. This approach offers several benefits; firstly, the crosslinking reaction between poly(acrylic acid) (PAA) and diglycidyl ethers can be performed in aqueous solution without the need for solvent exchange or a catalyst. Secondly, ‘greener’ routes towards the synthesis of acrylic acid are currently being investigated.9,10 Despite exhibiting remarkable elasticity and ductility, PAA–diglycidyl ether gels suffer from poor strength, significantly hindering practical applicability. It should also be noted that diglycidyl ether is very toxic, however there are derivatives, for example 1,6-heaxanediol diglycidyl ether (16DGE), that can be substituted which possess a minimal health risk.
In order to enhance spinning dope and filament strength, a small fraction of suitable filler can be compounded with the crosslinked polymer. Nanofibrillated cellulose (NFC) is an ideal choice for several reasons; it possesses a high stiffness (140–220 GPa) and aspect ratio (4–20 nm wide, 500–2000 nm long),11 while its effectiveness as a reinforcing material is well documented. Furthermore, its hydrophilic nature allows for dispersion in water and favourable compatibility with PAA.
As is often the case with polymeric materials, the incorporation of stiff reinforcement enhances strength at the expense of ductility and elasticity. The addition of plasticisers is not ideal, since they often negate the strength enhancement imparted by the filler. Furthermore, traditional polar plasticisers, such as glycerol12 or poly(ethylene glycol), can leach from their matrices and adsorb water molecules that can weaken the material. One viable approach involves compounding the filaments with a minimal amount of graphene or graphene oxide, two materials that have attracted significant attention as a novel lubricant,13 while also showing promise in cellulose-based composites.14,15 Furthermore, no attempts to prepare filaments from PAA–DGE gel systems, or enhance gel strength through compounding have been reported.
Herein, the preparation and characterisation of PAA–16DGE monofilaments compounded with NFC and reduced graphene oxide (RGO) is presented. Spinning dopes were prepared in aqueous solution, from which monofilaments were drawn. The influence of PAA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :crosslinker ratio and the concentration of NFC–RGO on filament mechanical, thermal and morphological properties were characterised.
:crosslinker ratio and the concentration of NFC–RGO on filament mechanical, thermal and morphological properties were characterised.
Experimental
Materials
Poly(acrylic acid) (Mw: ∼450000 g mol−1), graphite flakes (particle size + 100 mesh, ≥75% min−1), sulfuric acid (≥95%), hydrochloric acid (37%), potassium permanganate (99+%), sodium nitrate (99.5%), and hydrazine hydrate were purchased from Sigma Aldrich, USA. 1,6-Hexanediol diglycidyl ether was obtained from SACHEM Inc., Netherlands. Ammonia solution (28%) was purchased by VWR Co. Hydrogen peroxide (30%) was obtained from Merck. Nanofibrillated cellulose (NFC) suspension (1.39% wt) was provided by UPM Corporation (Helsinki, Finland). The NFC fibrils were mostly 20–30 nm in diameter and several micrometers in length. The material was manufactured by mechanical disintegration of bleached birch pulp, which was pre-treated with a Voith refiner prior to fibrillation with an M7115 fluidizer from Microfluidics Corp (Newton, MA, USA).16
NFC–RGO suspension preparation
Graphene oxide (GO) was prepared using a modified Hummers method.17 NFC–RGO suspension containing 5% wt RGO solid mass was prepared using a modified method.18 Briefly, GO dispersion with a solid content of 2 mg mL−1 was added to the NFC suspension (1.67% wt) and mechanically stirred at room temperature for 30 min under nitrogen flow. Deionised (DI) water was added to the mixture to lower the viscosity of the suspension. The final solid content of the NFC–GO mixture was 6.86 mg mL−1. The pH of the mixture was then adjusted to 10 using ammonia solution (25% wt). In the reduction step, hydrazine solution was injected into the reactor (100 μL of hydrazine per 100 mg of solid GO), which was then heated to 95 °C for 2 h. The effective reduction of GO to RGO by hydrazine could be observed by the color changing from yellow to dark black. The mixture was finally washed with DI water via vacuum filtration and re-dispersed in DI water to obtain the NFC–RGO suspension (1.39% wt solid mass, of which 5% wt is RGO).Spinning dope preparation
Appropriate amounts of 1,6-hexanediol diglycidyl ether and the NFC–RGO suspension were added to a 7% wt aqueous solution of PAA and stirred at room temperature for 1 h. The suspension was transferred to a 90 °C oven to allow the esterification (crosslinking) reaction to occur and concentrate the suspension to a suitable viscosity for filament spinning. The esterification reaction is summarized in Scheme 1. The suspension was subjected to manual stirring every 10 min to prevent film formation. The concentration was continued until filaments could be smoothly extruded without the wet filament adhering to the extrusion tip. An example of this spinning dope is presented in Fig. 1. | ||
| Scheme 1 Ester linkage (crosslink) formation between PAA and 16DGE. | ||
 | ||
| Fig. 1 Spinning dope of PAA–16DGE (1:1 ratios, no cellulose or graphene). | ||
Preparation of PAA–DGE–NFC–RGO filaments
The PAA–DGE–NFC–RGO monofilaments were prepared at room temperature. The suspensions were first charged into a 1 mL syringe equipped with a 20 gauge hypodermic needle (Terumo Europe NV, Belgium), having an inner diameter of 0.6 mm and tip length of 70 mm. Monofilaments of approximately 20 cm in length were injected by hand. Wet monofilaments were manually collected, attached vertically from both ends to paper strips, and dried under ambient conditions overnight. Residual moisture was removed by final drying at 120 °C for 10 min. A prepared monofilament is presented in Fig. 2, while the composition and nomenclature of the monofilaments are summarized in Table 1. | ||
| Fig. 2 PF11-2.5 monofilament. The square grids are approximately 7 × 7 mm2 in dimension. | ||
Characterisation of PAA–DGE–NFC–RGO filaments
Results and discussion
Monofilament structure and chemistry
:16DGE are presented in Fig. 3a. Amongst the various peaks within the pure PAA spectrum, the one of greatest interest is that of the carboxyl carbon at 184.7 ppm.19 The spectrum of PAA:16DGE also exhibits a peak at 184.7 ppm, however a more prominent peak appears at 180.0 ppm. This new peak is attributed to the ester carbons formed during crosslinking between PAA and 1,6-hexanediol diglycidyl ether, suggesting the reaction proceeded successfully.
 | ||
| Fig. 3 13C-NMR spectra; (a) pure PAA, PAA–16DGE filament, (b) selected PAA–16DGE–NFC–RGO monofilaments. | ||
The 13C NMR spectra of the PAA–DGE–NFC–RGO monofilaments are presented in Fig. 3b. As anticipated, the spectra displayed two peaks of varying intensity within the 180–190 ppm, corresponding to the unreacted carbonyl carbons of PAA (at higher ppm) and the ester carbons formed during crosslinking (at lower ppm). Monofilament PF11-2.5 displayed a sharp tall peak at 178.5 ppm, while a lower-intensity shoulder peak appeared at 185.5 ppm. This suggests a high degree of ester linkage formation (crosslinking) and relatively low amount of vacant carboxyl groups on the PAA chain. Increasing the PAA:16DGE ratio to 3:1 reduces the number of potential ester linkages formed between PAA and 1,6-hexanediol diglycidyl ether. Subsequently, specimen PF31-5 displayed two distinct peaks at 180.1 and 185.0 ppm, with the former exhibiting a slightly higher intensity. Several peaks attributed to NFC produce similar chemical shifts as those of 1,6-hexanediol diglycidyl ether, as shown in Fig. 3a. The potential of the crosslinker peaks to mask those of NFC, coupled with the relatively low concentrations of cellulose nanofibrils may be attributed to the lack of distinct peaks attributed to NFC.
In order to determine the extent of crosslinking (ester formation) within the monofilaments, the ratios of integrated peaks at 184.7 (13C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak of carboxylic acid) and 180.0 ppm (13CO peak of ester) were compared. These values are summarized in Table 2. The baseline 13CO (carboxyl):13CO (ester) ratio of PAA was 1.94 and was defined as 0% relative ester content. PAA–16DGE filament (containing no NFC or RGO) displayed an ester conversion value of 79%, indicating the monofilament contained unreacted, residual 1,6-hexanediol diglycidyl ether. PF11-2.5 displayed a conversion value of 91%, while PF11-5 exhibited a value of 88%. The increased degree of ester formation may be attributed to the possibility of esterification side reactions between PAA carboxyl groups and NFC hydroxyl groups.20 As anticipated, monofilaments with a PAA:16DGE ratio of 3:1 yielded lower ester conversion values than their 1:1 counterparts, due to the reduced concentration of 16DGE able to contribute towards ester formation. However, both PF31-2.5 and PF31-5 yielded similar values to PAA–16DGE, strongly suggesting the occurrence of additional esterification between NFC and PAA.
O peak of carboxylic acid) and 180.0 ppm (13CO peak of ester) were compared. These values are summarized in Table 2. The baseline 13CO (carboxyl):13CO (ester) ratio of PAA was 1.94 and was defined as 0% relative ester content. PAA–16DGE filament (containing no NFC or RGO) displayed an ester conversion value of 79%, indicating the monofilament contained unreacted, residual 1,6-hexanediol diglycidyl ether. PF11-2.5 displayed a conversion value of 91%, while PF11-5 exhibited a value of 88%. The increased degree of ester formation may be attributed to the possibility of esterification side reactions between PAA carboxyl groups and NFC hydroxyl groups.20 As anticipated, monofilaments with a PAA:16DGE ratio of 3:1 yielded lower ester conversion values than their 1:1 counterparts, due to the reduced concentration of 16DGE able to contribute towards ester formation. However, both PF31-2.5 and PF31-5 yielded similar values to PAA–16DGE, strongly suggesting the occurrence of additional esterification between NFC and PAA.
| Filament name | Integration ratio: 13CO (acid)/13CO (ester) |
Relative 13CO (ester) formation (%) |
Water content (% wt) |
|---|---|---|---|
| PAA | 1.94 | — | — |
| PAA–16DGE 1:1 |
0.41 | 79 | — |
| PF11-2.5 | 0.17 | 91 | 30 |
| PF11-5 | 0.16 | 88 | 17 |
| PF31-2.5 | 0.39 | 80 | 15 |
| PF31-5 | 0.37 | 81 | 9 |
:DGE ratio of 1:1 yielded slightly higher 920 cm−1 peak intensities than those with a 3:1 ratio, attributed to the difference in crosslinker concentration. The failure of the 16DGE to completely react with PAA carboxyl groups may be due to cellulose nanofibrils acting as barriers to ester linkage formation. Alternatively, esterification reactions between PAA and NFC may also have occurred.
 | ||
| Fig. 4 ATR-FT-IR spectra of 16DGE, PAA and PAA–16DGE–NFC–RGO monofilaments. | ||
As displayed in Fig. 4b, a broad peak at 3064 cm−1 within the pure PAA spectrum was attributed to O–H stretching of the carboxylic groups, while a shoulder at 3408 cm−1 corresponds to hydrogen-bonded water.22 Within filaments with a 1:1 PAA:16DGE ratio, the O–H stretching carboxyl peak at 3064 cm−1 disappeared, while in the 3:1 monofilaments the intensity was reduced. Additionally, the carboxylic CO stretch at 1693 cm−1 shifted to higher wavenumbers (Fig. 4c), the shift being greater in PAA:16DGE 1:1 monofilaments. Similar behaviour observed by Paralikar et al.23 was attributed to the formation of carboxylic esters.
The peak at 3408 cm−1 correlating to bound water displayed the greatest intensity for both 1:1 PAA:16DGE monofilaments. Similarly, PF31-2.5 exhibited a more-intense peak at 3408 cm−1 compared with PAA, while PF31-5 yielded an intensity similar to that of pure PAA. Peak intensity also decreased with increasing NFC/graphene concentration. This indicates that monofilament water content increases with crosslinking density and reduced at higher cellulose loadings.
Internal water content
To complement the FTIR data, the water content of the monofilaments were quantified gravimetrically; these results are summarised in Table 2. PF11-2.5 displayed an internal water content of 30%, which reduced to 17% upon increasing the NFC concentration to 5% wt. Similar results were observed within the 3:1 PAA:DGE filaments, with the water content reducing from 15 to 9% with increased cellulose loading. The water retention capacity of PAA is well known.20,24 The increased volume of cellulose nanofibrils within the monofilaments can act as barriers to interactions between PAA and water, reducing the potential of absorption. NFC may also reduce swelling capacity of the PAA:16DGE dopes, due to the reduced free volume within the crosslinked systems.
The PAA:16DGE ratio also had an evident effect on internal water content, reducing as the amount of PAA within the filaments increased. This behaviour may derive from the influence of crosslinker concentration on swelling capacity. Kohestanian and Bouhendi6 and Chen and Tan25 both observed increased swelling within poly(acrylic acid)-based gel systems as the crosslinker concentration was increased. At low crosslinker concentrations, adsorbed water is unable to be maintained within the polymer gels due to low crosslinking density. As the crosslinker concentration (and crosslink density) is increased, the swelling ability and potential amount of retained adsorbed water increases.
Furthermore, the increased water content at higher crosslinker concentration may derive from possible side reactions that 1,6-hexanediol can partake in. In addition to the esterification reaction that occurs between 16DGE and PAA, water and vacant hydroxyl groups within the system can react with 16DGE. These reaction are summarised in Scheme 2. The product of these reactions contain numerous hydroxyl groups which can readily adsorb water molecules, or further react with carboxyl moieties on PAA to yield water as a by-product. At increase 16DGE concentrations, the likelihood of these reactions occurring is significantly increased. However, the FTIR and 13C NMR data indicate that ester formation predominates within the filaments.
 | ||
| Scheme 2 Possible side-reactions which can occur within the PAA–16DGE–NFC–RGO monofilaments. Adapted from Kohestanian and Bouhendi.6 | ||
Surface morphology
Scanning electron micrographs of selected PAA–16DGE–NFC–RGO filaments are presented in Fig. 5. Monofilament PF11-2.5 (Fig. 5a) displayed a smooth, uniform morphology with an average diameter of ∼80 μm. The bulk of the NFC–RGO is distributed within the filament, with no visible agglomerates or layers protruding through the PAA–16DGE surface. Increasing the NFC concentration to 5% wt (and graphene conc. to 0.25% wt) (Fig. 5b) increased the average filament diameter to ∼110 μm, while increasing the surface roughness. This may be attributed to the increased solids content within the spinning dope, resulting in a greater amount of pressure required to spin the dope through the syringe. This equates to a greater shear rate experienced by the spinning dope. As the filament leaves the nozzle, the increased shear rate leads to a subsequent greater expansion within the filaments perpendicular to the spinning direction. This increase in jet swell results is monofilaments with larger diameters,26,27 while the surface roughness may derive from NFC–RGO segments protruding the filament surface during disorientation upon exiting the nozzle. | ||
| Fig. 5 Scanning electron micrographs of PAA–16DGE–NFC–RGO monofilaments; (a) PF11-2.5, (b) PF11-5. | ||
Furthermore, monofilaments PF11-5 exhibited small aggregates appearing on its surface. Hydrogen bonding between cellulose nanofibrils28 and van der Waals forces between graphene layers29 result in aggregate formation. It is most probable that cellulose aggregation occurred during the concentration of the spinning dope (conducted at 90 °C). As water was gradually removed from the dope and the PAA–DGE–NFC–RFO suspensions became more concentrated, the likelihood of interfibril hydrogen bonding via NFC's hydroxyl groups increases.
Thermal stability and degradation
The mass-loss curves of the monofilaments are presented in Fig. 6a. All materials exhibited degradation curves with similar characteristics; an initial decrease in mass begins at ∼50 °C and is attributed to the release of residual water within the filaments. A second degradation step follows at ∼200 °C during which anhydride-type structures form and subsequently decarboxylate.30 The third and major degradation event (starting temperature: ∼320 °C) involves chain scission and depolymerisation of PAA, resulting in the formation of short chain-fragments.31 | ||
| Fig. 6 TGA data of PAA–16DGE–NFC–RGO monofilaments; (a) mass-loss curve, (b) derivative mass-loss curve. | ||
The mass-loss rate curves are presented in Fig. 6b. Following the initial peak attributed to water elimination, a peak corresponding to anhydride formation and decarboxylation during the second degradation event is noticeable; the maximum of this peak (Td2) varied with monofilament composition. PF11-2.5 exhibited a Td2 of 279 °C, while the Td2 of PF11-5 reduced to 256 °C. PF31-2.5 and PF31-5 both displayed the lowest Td2 values of 238 °C. This is attributed to the degree of crosslinking within the monofilaments. During the second degradation event, an aldehyde linkage can be formed from two carboxylic acid moieties.32 Increasing the degree of crosslinking within PAA results in; (a) less vacant –COOH groups being able to partake in aldehyde formation and (b) more energy required to break ester bonds which may subsequently react to form anhydrides. Furthermore, when comparing the relative percentage of ester formation (crosslinking) calculated from the 13C NMR data (refer Table 2), the order of ester formation is PF11-2.5 > PF11-5 > PF31-2.5 ≈ PF31-5. This order is identical to that of the Td2 values.
When comparing the peak maximum of the third degradation event (Td3), all monofilaments displayed maxima at ∼422 °C (within ±2 °C). However, the onset of this degradation event occurred at much lower temperatures for PF31-2.5 and PF31-5 (both 279 °C) than PF11-25 and PF11-5 (both 339 °C). This again can be attributed to the increased energy required for bond scission and depolymerisation within more crosslinked filaments.
PAA–16DGE 1:1 (containing no NFC or RGO) displayed a Td3 values of 422 °C, within the same range as the monofilaments, and a Td2 value of 242 °C. This is close to the Td2 value of 238 °C recorded for PF31-2.5 and PF31-5. Considering the relative ester formation percentage of PAA–16DGE 1:1 was 79% (again, close to the 80 and 81% recorded for PF31-25 and PF31-5, respectively), it is proposed that the primary factor influencing the second and third degradation steps is the degree of crosslinking. However, the peak temperature corresponding to water loss (Td1) of PAA–16DGE 1:1 was 108 °C, considerably lower than Td1 value of 144 °C exhibited by all monofilaments. Although the water content of the monofilaments varied with composition (refer Table 2), the peak temperature Td1 remained the same irrespective of NFC–RGO concentration. Furthermore, the fact that a difference in Td1 was observed for PAA–16DGE 1:1, PF11-2.5 and PF11-5 (all containing the same PAA:16DGE ratio), it is proposed that the increase in Td1 is due to NFC and RGO creating ‘most tortuous path’ for water molecules during heating. Hydrogen bonding between water molecules and NFC or RGO can also increase water retention, requiring more energy to eliminate water and subsequently increasing Td1.
Tensile properties
The stress–strain curves of the various filaments are presented in Fig. 7, while the tensile data is summarised in Table S1 of the ESI.† PAA–16DGE monofilament (PAA:16DGE % wt ratio 1:1) exhibited an initial modulus of 2.1 MPa, tensile strength of 3.9 MPa, strain at break of 411% and work-to-fracture of 986.3 MJ m−3. To demonstrate the reinforcing ability of cellulose nanofibrils, a PAA–DGE monofilament compounded with 5% wt NFC (no graphene) was prepared and characterised; as anticipated, the addition of nanocellulose significantly enhanced moduli (12.3 MPa), strength (20.8 MPa) and work-to-fracture (1870.0 MJ m−3) values, while also drastically reducing elongation (135%). When comparing specimen PF11-2.5, the initial modulus was 4.7 MPa. However, tensile strength increased to 45.5 MPa while the elongation at break was 486%. Furthermore, the work-to-fracture of specimen PF11-2.5 drastically increased to 9235.2 MJ m−3, more than nine times higher than pure PAA–16DGE 1:1.
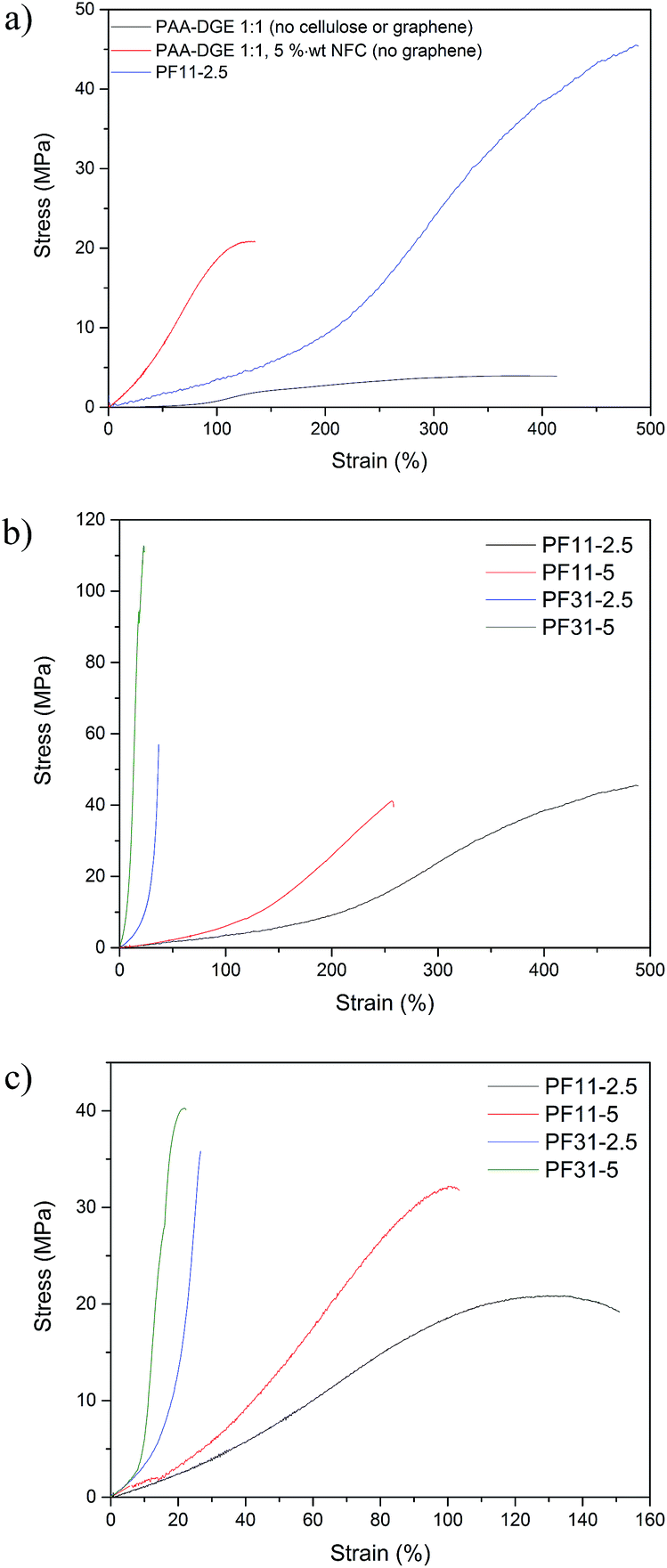 | ||
| Fig. 7 Stress–strain curves of selected monofilaments; (a) highlighting the influence of NFC and graphene, (b) ‘dry’ PAA–16DGE–NFC–RGO specimens, (c) PAA–16DGE–NFC–RGO specimens following immersion in water. | ||
These unprecedented mechanical properties stem from two primary factors; (1) the influence of cellulose nanofibrils and (2) the influence of graphene. Firstly, the incorporation of NFC imparts stiffness into the filaments and allows for transfer of stress during moments of applied load. The reinforcement ability of NFC is well documented and is facilitated primarily via hydrogen bonding between PAA and the cellulose nanofibrils. It is possible that covalent bonding may also occur between NFC and PAA20 or NFC and 16DGE,6 encouraging further stress transfer and reinforcing ability.
Secondly, the lubricating influence of graphene reduces the interfibril friction of NFC, the primary factor that determines elongation and ductility.33 Graphene layers coat the NFC, allowing them to experience interfibril slippage and possible alignment in the deformation direction. A proposed mechanism of this lubricating effect is presented in Scheme 3. In aqueous solution, ordered cellulose chains and graphene can form a stable complex, generally interacting via strong hydrogen bonding although CH–π interactions are also possible.34 Cellulose most closely interacts with graphene along its uniplanar hydrophobic face, with water being excluded to the outer edges of cellulose. Since water minimizes the friction of graphene,35 interaction between a hydrophobic graphene layer and hydrophilic cellulose edge containing bound water results in slippage of the respective layers past one another. Strong interactions between cellulose and graphene and/or graphene oxide are also known to contribute towards the mechanical reinforcement of cellulosic36 and PAA-based37 filaments, therefore the possibility of graphene contributing (albeit, a minor contribution) towards reinforcing the PAA–DGE filaments should not be totally excluded.
 | ||
| Scheme 3 The lubricating effect of graphene within PAA–16DGE–NFC–RGO monofilaments; (a) under zero strain, (b) during applied load. The dashed lines represent hydrogen bonding. | ||
The influence of PAA:DGE ratio and NFC–RGO concentration on the tensile properties are presented in Fig. 7b. Increasing the NFC concentration from 2.5 to 5% resulted in increased moduli and strength values, while reducing elongation. This was indicative of the reinforcing ability of cellulose nanofibrils. The additional likelihood of NFC–PAA interactions and possible NFC–DGE side reactions is increased with cellulose loading. However, the drastic reduction in elongation compare with filaments containing 2.5% wt NFC suggests that the increased volume of NFC significantly hinder the sliding of PAA chains during deformation.
Increasing the PAA:DGE ratio from 1:1 to 3:1 (that is, increasing the PAA concentration within the filaments) resulted in stiffer monofilaments, exemplified by increased moduli and strength values and diminished elongation at break. This is in contrast to the expected increase in strength and modulus associated with increasing crosslinker concentration within crosslinked polymer systems. This may be attributed to the observed increase in swelling capacity of the spinning dopes PAA:DGE ratios of 1:1 (refer Table 2). As a result, filaments containing a PAA:DGE ratio of 1:1 have a greater degree of crosslinking and increased likelihood of adsorbed water following drying. Furthermore, the increased water content may have a greater impact on reducing the surface friction of graphene, enhancing its lubricating ability and resulting in enhanced elongation.
Wet-strength properties
The stress–strain curves of the PAA–DGE–NFC–RGO filaments following water immersion are presented in Fig. 7c. Exposure to water had an evident impact on tensile properties; for filaments with a PAA:DGE ratio of 1:1, the reduction in elongation was most drastic, while the strength and moduli values reduced somewhat less severely. In contrast, PAA:DGE 3:1 filaments experienced a negligible impact on elongation and moduli, while the strength was more significantly impacted. This was especially the case for filament PF31-5. This deterioration in filament tensile properties and structural integrity can be attributed to the superabsorbent nature of PAA. The chain swelling and softening of PAA that follows results in reduced polymer chain integrity under applied load.
Increasing the NFC–RGO concentration had the same effect on tensile behaviour for both the water-immersed and ‘dry’ set of filaments. This indicates that NFC continued to reinforce the PAA:DGE filaments following immersion in water. It is also likely that water molecules may preferentially adhere to superabsorbent PAA chains, rather than disrupting NFC–NFC and/or NFC–PAA hydrogen bonding. These results highlight the robustness of NFC as a reinforcement material while also reiterating that filament water stability is ultimately influenced by PAA.
Conclusions
PAA–16DGE–NFC–RGO monofilaments were successfully spun from aqueous solution. Crosslinking between PAA and 16DGE in the form of ester linkages was confirmed via 13C-NMR and ATR-FT-IR. The degree of crosslinking increased with crosslinker concentration, while PAA–NFC ester linkage formation was also a likely occurrence. Increased crosslinking density also increased the likelihood of water molecules being retained by the filaments, thus increasing the internal water content. Subsequently, 1:1 PAA:16DGE ratios resulted in softer, more ductile and elastic filaments due to the higher internal water content. Thermal degradation and stability were influenced by both NFC/RGO and the degree of crosslinking; the former retarded the extent of water elimination, while the latter dictated the extent of aldehyde formation, chain scission and depolymerisation of PAA.
Sufficient interaction between cellulose nanofibrils and the PAA matrix, encouraged due to their polar chemical structures, allowed NFC to reinforce the monofilaments. This was displayed via enhanced moduli and strength values. High elongation values were maintained within monofilaments, which was attributed to the unique synergy between NFC and graphene; under moments of applied load, layers of graphene lubricate cellulose nanofibrils, allowing them experience interfibril slippage and possible alignment. Tensile properties diminished following exposure to water, due to material softening associated with excessive water uptake. The influence of NFC–RGO and 16DGE concentration was identical for both ‘dry’ monofilaments and those exposed to water, reinforcing the influence these components exert on monofilament performance.
These hybrid monofilaments possess great potential due to their simple, water-based synthesis, renewable and sustainable raw materials and high ductility, elasticity and work-to-break. The promising initial results encourage further development of a new class of ductile, elastic soft materials, with potential applications in textile, homeware and consumer product markets.
Acknowledgements
The authors wish to thank UPM Corporation for providing the nanofibrillated cellulose and SACHEM Inc. for providing the 1,6-hexanediol diglycidyl ether.Notes and references
- F. Hämmerle, Lenzinger Ber., 2011, 89, 12–21 Search PubMed.
- L. Shen and M. Patel, Lenzinger Ber., 2010, 88, 1–59 CAS.
- S. Zikeli, US Patent 5,603,883, 1997.
- Aalto University, From cellulose to textile fiber, ScienceDaily, 27 November 2013.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- M. Kohestanian and H. Bouhendi, Colloid Polym. Sci., 2015, 1–13 Search PubMed.
- M. Dauben, K. Reichert, P. Huang and J. Fock, Polymer, 1996, 37, 2827–2830 CrossRef CAS.
- U. Mähr, H. Purnama, E. Kempin, R. Schomäcker and K. Reichert, J. Membr. Sci., 2000, 171, 285–291 CrossRef.
- B. Yan, L. Tao, Y. Liang and B. Xu, ACS Catal., 2014, 4, 1931–1943 CrossRef CAS.
- B. Yan, L. Tao, Y. Liang and B. Xu, ChemSusChem, 2014, 7, 1568–1578 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- A. Jones, M. Zeller and S. Sharma, Progress in Biomaterials, 2013, 2, 12 CrossRef.
- D. Berman, A. Erdemir and A. V. Sumant, Mater. Today, 2014, 17, 31–42 CrossRef CAS.
- N. D. Luong, N. Pahimanolis, U. Hippi, J. T. Korhonen, J. Ruokolainen, L. Johansson, J. Nam and J. Seppala, J. Mater. Chem., 2011, 21, 13991–13998 RSC.
- B. Wang, W. Lou, X. Wang and J. Hao, J. Mater. Chem., 2012, 22, 12859–12866 RSC.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Osterberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- M. U. M. Patel, N. D. Luong, J. Seppälä, E. Tchernychova and R. Dominko, J. Power Sources, 2014, 254, 55–61 CrossRef CAS.
- L. Nguyen Dang and J. Seppälä, Cellulose, 2015, 22, 1799–1812 CrossRef.
- S. Maeda, Y. Fujiwara, C. Sasaki and K.-K. Kunimoto, Polym. J., 2011, 44, 200–203 CrossRef.
- S. Spoljaric, A. Salminen, L. Nguyen and J. Seppälä, Cellulose, 2013, 20, 2991–3005 CrossRef CAS.
- M. González, J. Cabanelas and J. Baselga, Applications of FTIR on Epoxy Resins - Identification, Monitoring the Curing Process, Phase Separation and Water Uptake, in Infrared Spectroscopy - Materials Science, Engineering and Technology, ed. T. Theophanides, InTech, 2012, pp. 261–284 Search PubMed.
- T. Kondo, Cellulose, 1997, 4, 281–292 CrossRef CAS.
- S. A. Paralikar, J. Simonsen and J. Lombardi, J. Membr. Sci., 2008, 320, 248–258 CrossRef CAS.
- A. Pourjavadi, R. Soleyman and G. R. Barajee, Starch–Stärke, 2008, 60, 467–475 CrossRef CAS.
- Y. Chen and H. Tan, Carbohydr. Res., 2006, 341, 887–896 CrossRef CAS PubMed.
- E. Morris, Bench-scale, Multifilament Spinning Conditions Effect on the Structure and Properties of Polyacrylonitrile Precursor Fiber, M.Sc thesis, University of Kentucky, 2011, p. 74.
- G. Peng, Y. Wen, Y. Yang and L. Liu, Int. J. Polym. Anal. Charact., 2008, 13, 369–375 CrossRef CAS.
- K. Ali Chami and R. Sylvain, J. Nanomater., 2013, 2013, 1–10 Search PubMed.
- Y. Wan, L. Tang, D. Yan, L. Zhao, Y. Li, L. Wu, J. Jiang and G. Lai, Compos. Sci. Technol., 2013, 82, 60–68 CrossRef CAS.
- S. Dubinsky, G. S. Grader, G. E. Shter and M. S. Silverstein, Polym. Degrad. Stab., 2004, 86, 171–178 CrossRef CAS.
- I. C. McNeill and S. M. T. Sadeghi, Polym. Degrad. Stab., 1990, 29, 233–246 CrossRef CAS.
- J. J. Maurer, D. J. Eustace and C. T. Ratcliffe, Macromolecules, 1987, 20, 196–202 CrossRef CAS.
- M. Wang, A. Olszewska, A. Walther, J. Malho, F. H. Schacher, J. Ruokolainen, M. Ankerfors, J. Laine, L. A. Berglund, M. Osterberg and O. Ikkala, Biomacromolecules, 2011, 12, 2074–2081 CrossRef CAS PubMed.
- R. Alqus, S. J. Eichhorn and R. A. Bryce, Biomacromolecules, 2015, 16, 1771–1783 CrossRef CAS PubMed.
- E. Singh, A. V. Thomas, R. Mukherjee, X. Mi, F. Houshmand, Y. Peles, Y. Shi and N. Koratkar, ACS Nano, 2013, 7, 3512–3521 CrossRef CAS PubMed.
- Y. Li, H. Zhu, S. Zhu, J. Wan, Z. Liu, O. Vaaland, S. Lacey, Z. Fang, H. Dai, T. Li and L. Hu, NPG Asia Mater., 2015, 7, e150 CrossRef CAS.
- Z. Jiang, Q. Li, M. Chen, J. Li, J. Li, Y. Huang, F. Besenbacher and M. Dong, Nanoscale, 2013, 5, 6265–6269 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table summarizing the tensile data. See DOI: 10.1039/c5ra24539g |
| This journal is © The Royal Society of Chemistry 2015 |