
Slow photocharging and reduced hysteresis in low-temperature processed planar perovskite solar cells

Abstract
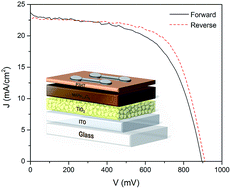
High efficient organolead trihalide perovskite solar cells typically utilize high temperature processed TiO2 as the electron transporting layer (ETL). A new method for the preparation of low temperature processed amorphous TiO2 as the ETL in planar perovskite solar cells is demonstrated. The proposed technique allowed the entire fabrication of perovskite solar cells with power conversion efficiencies as high as 13.7%, to take place at temperatures lower than 150 °C. We revealed that the source of device efficiency lies in the fast and balanced charge transport within the selective contacts as well as the effective charge extraction from the perovskite.
 Please wait while we load your content...
Please wait while we load your content...

