
Synthesis of isatins by the palladium-catalyzed intramolecular acylation of unactivated aryl C(sp2)–H bonds†
Jue Li,
Yang Zheng,
Xinling Yu,
Songyang Lv,
Qiantao Wang,
Li Hai* and
Yong Wu*
Key Laboratory of Drug-Targeting of Education Ministry and Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu, 610041, P. R. China. E-mail: wyong@scu.edu.cn; smile@scu.edu.cn; Fax: +86 02885506666
First published on 27th November 2015
Abstract
Synthesis of isatins from formyl-N-arylformamides is achieved via PdCl2-catalyzed intramolecular acylation. This method shows the possibility of Pd-catalyzed aryl C(sp2)–H bond activation on the synthesis of isatins, affording an array of isatins in good yields. Yet this protocol is operationally simple and atom economical.
Over the last decade, transition-metal-catalyzed C–H bond functionalization has emerged as a useful and powerful synthetic strategy, and has allowed chemist to assemble complex molecular structures efficiently. Metals such as Pd, Cu, Ni, Fe, Ru, etc. play vital roles on the transformation of unactivated C(sp2)–H bond to C(sp2)–C bond with the assistance of directing group.1 In particular, the Pd-catalyzed direct C–H bond acylation protocols using aryl compounds and some simple precursors have been widely investigated,2 such as the Pd catalyzed ortho-aroylation using benzyl bromides as aroyl surrogates,2a the decarboxylative acylation of arenes with mandelic acid derivatives via palladium catalyst,2b and the Pd-catalyzed oxidative C–H bond acylation of acetanilides employing toluene derivatives as the coupling precursors.2c These methods provide efficient pathway to afford aryl ketone motifs.
On the other hand, the isatin derivatives, which have similar structures with aryl ketone motifs, have received much attention, due to their biological activity and potential medical value3 and the activities of C-3 carbonyl group as a synthetic intermediate.4 Traditionally, Sandmeyer procedure,5 Stollé procedure6 and Martinet procedure7 are used to synthesize isatin. However, these methods suffer from the harsh conditions, poor yields and limited substrate choices. This motivates the development of new synthetic methods to overcome these limitations. Inspiringly, a number of modern and efficient methods have been exploited, for example, the ylide-mediated carbonyl homologation of anthranilic acids,8 and the I2-mediated9 or Cu-catalyzed10 intramolecular cyclic amidation from ortho-substituted anilines. Although these reactions showed some improvements, the requirement of the ortho-functionalized aromatic substrates, which were not readily available, were the obvious drawbacks. In contrast to preparation of ortho-functionalized substrates, the direct transformation from sp2 C–H bonds of aryl moiety to C–C bonds for the synthesis of isatin is a more appealing alternative approach.
In a recent J. Am. Chem. Soc. communication, Li and co-workers have reported intramolecular acylation of formyl-N-arylformamides 1 to indoline-2,3-diones 2 based on Cu-catalyzed activation of C–H bond of aldehyde[Scheme 1a, eqn (1)].11 Their work was interesting as it simplified the substrate preparation and revealed the possibility of intramolecular acylation. They suggested that the C–H bond on aldehyde can be activated by CuCl2 with O2 in Schlenk tube at 100 °C. Notably, they also mentioned that some palladium complexes, including Xiao's catalytic system [Pd(dba)2 and dppp in DMF at 115 °C],12 Martin's catalytic system [Pd(OAc)2 and rac-BINAP in dioxane at 110 °C]13 and Cheng's catalytic system [Pd(OAc)2 in xylene at 120 °C],14 are not compatible to this reaction [Scheme 1a, eqn (2)]. This, however, is in contrary with the well accepted Pd-catalyzed ortho-acylation of acetanilides.2c,2h,i Considering formyl-N-arylformamide 1 is a acetanilide derivative, it is possible that unactivated aryl C(sp2)–H bond of compound 1 would be activated via suitable Pd catalyst and intramolecular acylation could happen, upon which isatins 2 could be obtained (Scheme 1b). So we carefully reviewed Li and co-workers' work, and repeated the experiments with conditions mentioned above. N-Methyl-2-oxo-N-phenylacetamide 1a, which we had previously synthesized, was employed as the substrate (Table 1, entry 1–3).15 Not surprisingly, all reactions suffered from poor yield (6–13%) although the reaction facilitated by Pd(OAc)2 in xylene had a relatively higher yield (13%). However, replacing Pd(OAc)2 by PdCl2 in the later reaction, the yield of 2a was increased to 61% (Table 1, entry 4). This encouraged us as it implies that there might be a possibility to further improve the reaction. By our best knowledge, no previous reports have described such intramolecular acylation of acetanilide derivatives via Pd-activated aryl C(sp2)–H bond (Scheme 1b), therefore, we explored more about it.
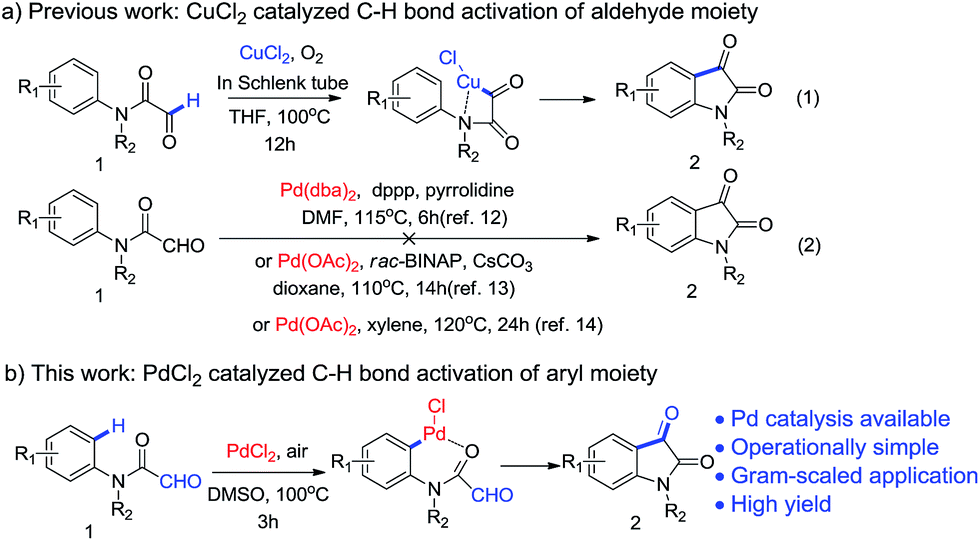 | ||
| Scheme 1 Synthesis of isatins using Pd catalyst. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), catalyst (as indicated), solvent (2 ml) and additive (as indicated) were heated for indicated reaction time.b Isolated yield. BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; TFA = trifluoroacetate; dba = dibenzylideneacetone; PPh3 = triphenylphosphine; dppf = 1,1′-bis(diphenylphosphino)ferrocene; dppp = 1,3-bis(diphenylphosphino)propane. | ||||||
| 1 | Pd(OAc)2 (10) | Cs2CO3/BINAP/air | Dioxane | 110 | 14 | 6 |
| 2 | Pd(dba)2 (10) | Pyrrolidine/dppp/air | DMF | 115 | 6 | 11 |
| 3 | Pd(OAc)2 (10) | Air | Xylene | 120 | 24 | 13 |
| 4 | PdCl2 (10) | Air | Xylene | 120 | 24 | 61 |
| 5 | PdCl2 (10) | Air | Xylene | 100 | 3 | 70 |
| 6 | PdCl2 (10) | Air | Dioxane | 100 | 3 | 76 |
| 7 | PdCl2 (10) | Air | DMF | 100 | 3 | 65 |
| 8 | PdCl2 (10) | Air | Toluene | 100 | 3 | 73 |
| 9 | PdCl2 (10) | Air | DMSO | 100 | 3 | 92 |
| 10 | Pd(TFA)2 (10) | Air | DMSO | 100 | 3 | 90 |
| 11 | Pd(PPh3)4 (10) | Air | DMSO | 100 | 3 | <5 |
| 12 | PdCl2(dppf)·CH2Cl2 (10) | Air | DMSO | 100 | 3 | 33 |
| 13 | PdCl2(PPh3)2 | Air | DMSO | 100 | 3 | <5 |
| 14 | — | Air | DMSO | 100 | 3 | — |
| 15 | PdCl2 (5) | Air | DMSO | 100 | 3 | 78 |
| 16 | PdCl2 (20) | Air | DMSO | 100 | 3 | 92 |
| 17 | PdCl2 (100) | Argon | DMSO | 100 | 3 | 18 |
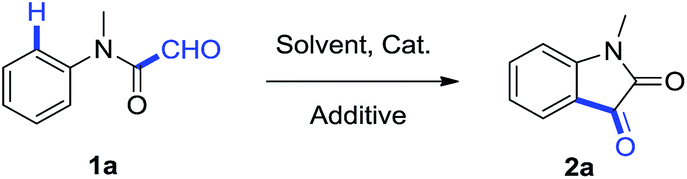
In beginning of our study, side products were detected when the reaction ran at 120 °C for more than 24 hours (Table 1, entry 4). After testing the reaction with different temperatures and different reaction time, it was found that the reaction gave a better yield at 100 °C after 3 h (Table 1, entry 5). Thus, later reactions were carried out at 100 °C for 3 h. Then we tested different solvents and found that DMSO was more effective than other solvents such as DMF, toluene and 1,4-dioxane (Table 1, entry 6–9). Furthermore, evaluation on other Pd compounds was carried out, and suggested that PdCl2 was still the most suitable catalyst (Table 1, entry 10–13). It is also found that 10 mol% PdCl2 was necessary as neither increasing nor decreasing the catalyst loading did not give a higher yield of 2a. No desired product was obtained without the presence of PdCl2 (Table 1, entry 14–16). Notably air served as the essential factor because the yield of 2a was unsatisfied when this reaction was run in argon atmosphere, even though the catalyst loading was up to 1 equiv. (Table 1, entry 17).
Accordingly, the reaction conditions are optimized as follows: 10 mol% PdCl2 under air in DMSO at 100 °C for 3 h.
After the reaction condition was optimized, the substrate scope of this synthetic methodology was also established and the results were shown in Table 2. Functional groups attached to the nitrogen atom, such as ethyl, n-butyl, allyl, gave the corresponding products in good yields (Table 2, compound 2a–d). Electron-donating and electron-withdrawing groups at 4th position of aromatic ring were well tolerated during the reaction (Table 2, compound 2f–l). Notably the substrates with meta-methyl or meta-halogens provided a mixture of 4-substituted and 6-substituted indoline-2,3-diones in different proportion (Table 2, compound 2m–o). Other functional groups, such as cyano, substituted phenyls, and heterocycle, were compatible with the optimal condition (Table 2, compound 2p–v). It was noteworthy that under the optimal condition the reaction can be easily scaled up, for example, when 6.1 mmol of 1a (1.0 g) was used, this reaction was showed to have similar efficiency when it was under the optimal condition (88%).

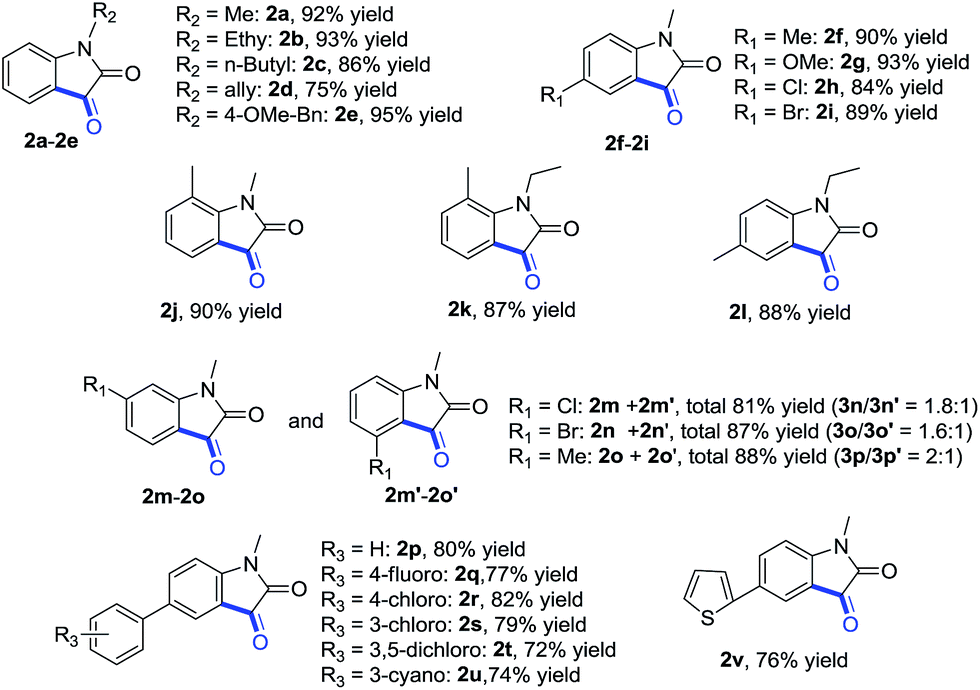
According to the previous studies,2,16 a plausible mechanism as shown in Scheme 2 was proposed. Firstly, intermediate 3a was formed through ortho-palladation of 1a. Then a carbopalladation reaction between the aryl-Pd(II) moiety and the formyl group would give the Pd(II) alkoxide intermediate 4a. Subsequently, the isatin 2a and Pd(0) could be obtained by β-hydride elimination. The Pd(0) was then oxidized to Pd(II), hence the regeneration of the Pd(II) catalyst, which fulfil the catalytic cycle.
 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusions
In conclusion, we have described an efficient synthesis of isatin derivatives via PdCl2-catalyzed intramolecular acylation of formyl-N-arylformamides using air as oxidant. This atom economical method implies that such a transformation could also be facilitated by a suitable Pd catalyst, albeit some reported palladium complexes did not work.12–14 This method offers not only good yields of the corresponding product, but also has a good tolerance with different functional groups. In addition, this protocol can be easily scaled up. Also, using air and DMSO as catalytic system makes this method operationally simple. This powerful tool that shows versatility and flexibility can be a good addition to the field of organic synthesis and medicinal chemistry.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (NSFC) (grant number 81373259).Notes and references
- For selected example, see: (a) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (b) I. Bauer and H. J. Knőlker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (c) X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (e) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Behera, W. Ali, S. Guin, N. Khatun, P. R. Mohanta and B. K. Patel, RSC Adv., 2015, 5, 33334 RSC; (b) X. Liu, Z. Yi, J. H. Wang and G. Y. Liu, RSC Adv, 2015, 5, 10641 RSC; (c) Y. N. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (d) Q. Zhang, F. Yang and Y. Wu, Chem. Commun., 2013, 49, 6837 RSC; (e) A. B. Khemnar and B. M. Bhanage, Eur. J. Org. Chem., 2014, 2014, 6746 CrossRef CAS; (f) S. Han, S. Sharma, J. Park, M. Kim, Y. Shin, N. K. Mishra, J. J. Bae, J. H. Kwak, Y. H. Jung and I. S. Kim, J. Org. Chem., 2014, 79, 275 CrossRef CAS PubMed; (g) S. Sharma, I. A. Khan and A. K. Saxena, Adv. Synth. Catal., 2013, 355, 673 CrossRef CAS; (h) J. Weng, X. Liu and G. Zhang, Tetrahedron Lett., 2013, 54, 1205 CrossRef CAS; (i) Y. N. Wu, B. Z. Li, F. Mao, X. S. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed; (j) P. Fang, M. Z. Li and H. B. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Zou, W. L. Chan, M. Ding, S. Y. Leong, S. Nilar, P. G. Seah, W. Liu, R. Karuna, F. Blasco, A. Yip, A. Chao, A. Susila, H. Dong, Q. Y. Wang, H. Y. Xu, K. Chan, K. F. Wan, F. Gu, T. T. Diagana, T. Wagner, I. Dix, P. Y. Shi and P. W. Smith, ACS Med. Chem. Lett., 2015, 6, 344 CrossRef CAS PubMed; (b) B. K. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, S. Keller-Maerki, C. Fischli, A. Goh, E. K. Schmitt, P. Krastel, E. Francotte, K. Kuhen, D. Plouffe, K. Henson, T. Wagner, E. A. Winzeler, F. Petersen, R. Brun, V. Dartois, T. T. Diagana and T. H. Keller, J. Med. Chem., 2010, 53, 5155 CrossRef CAS PubMed; (c) C. Cleva, J. F. Arrighi, J. Atherall, J. Macritchie, T. Martin, Y. Humbert, M. Gaudet, D. Pupowicz, M. Maio, P. A. Pittet, L. Golzio, C. Giachetti, C. Rocha, G. Bernardinelli, Y. Filinchuk, A. Scheer, M. K. Schwarz and A. Chollet, J. Med. Chem., 2008, 51, 2227 CrossRef PubMed; (d) G. Smith, M. Glaser, M. Perumal, Q. D. Nguyen, B. Shan, E. Årstad and E. O. Aboagye, J. Med. Chem., 2008, 51, 8057 CrossRef CAS PubMed; (e) G. Krishnegowda, A. P. Gowda, H. R. S. Tagaram, K. F. Staveley-O'Carroll, R. B. Irby, A. K. Sharma and S. Amin, Bioorg. Med. Chem., 2011, 19, 6006 CrossRef CAS PubMed; (f) T. M. Bridges, J. P. Kennedy, M. J. Noetzel, M. L. Breininger, P. R. Gentry, P. J. Conn and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2010, 20, 1972 CrossRef CAS PubMed.
- For selected examples, see: (a) J. F. Da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273 CrossRef CAS; (b) Y. Liu, H. Wang and J. Wan, Asian J. Org. Chem., 2013, 2, 374 CrossRef CAS; (c) M. A. Borad, M. N. Bhoi, N. P. Prajapati and H. D. Patel, Synth. Commun., 2014, 44, 897 CrossRef CAS; (d) P. Pakravan, S. Kashanian, M. M. Khodaei and F. J. Harding, Pharmacol. Rep., 2013, 65, 313 CrossRef CAS PubMed; (e) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed.
- T. Sandmeyer, Helv. Chim. Acta, 1919, 2, 234 CrossRef CAS.
- R. Stollé, Ber. Dtsch. Chem. Ges., 1913, 46, 3915 CrossRef.
- J. Martinet, C. R. Hebd. Seances Acad. Sci., 1918, 166, 851 CAS.
- C. T. Lollar, K. M. Krenek, K. J. Bruemmer and A. R. Lippert, Org. Biomol. Chem., 2014, 12, 406 CAS.
- (a) G. Satish, A. Polu, T. Ramar and A. Ilangovan, J. Org. Chem., 2015, 80, 5167 CrossRef CAS PubMed; (b) M. R. Reddy, N. N. Rao, K. Ramakrishna and H. M. Meshram, Tetrahedron Lett., 2014, 55, 4758 CrossRef; (c) A. Ilangovan and G. Satish, J. Org. Chem., 2014, 79, 4984 CrossRef CAS PubMed; (d) F. F. Gao, W. J. Xue, J. G. Wang and A. X. Wu, Tetrahedron, 2014, 70, 4331 CrossRef CAS.
- (a) J. Huang, T. Mao and Q. Zhu, Eur. J. Org. Chem., 2014, 2014, 2878 CrossRef CAS; (b) P. C. Huang, P. Gandeepan and C. H. Cheng, Chem. Commun., 2013, 49, 8540 RSC; (c) A. Ilangovan and G. Satish, Org. Lett., 2013, 15, 5726 CrossRef CAS PubMed; (d) T. Liu, H. Yang, Y. Jiang and H. Fu, Adv. Synth. Catal., 2013, 355, 1169 CrossRef CAS.
- B. X. Tang, R. J. Song, C. Y. Wu, Y. Liu, M. B. Zhou, W. T. Wei and J. H. Li, J. Am. Chem. Soc., 2010, 132, 8900 CrossRef CAS PubMed.
- J. W. Ruan, O. Saidi, J. A. lggo and J. L. Xiao, J. Am. Chem. Soc., 2008, 130, 10510 CrossRef CAS PubMed.
- P. Alvarez-Bercedo, A. Flores-Gaspar, A. Correa and R. Martin, J. Am. Chem. Soc., 2010, 132, 466 CrossRef CAS PubMed.
- X. F. Jia, S. H. Zhang, W. H. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120 CrossRef CAS PubMed.
- (a) Y. Zheng, Z. Zhan, X. Cheng, X. Ma, J. Li, L. Hai and Y. Wu, Asian J. Org. Chem., 2015, 4, 890 CrossRef CAS; (b) Z. Zhan, X. Cheng, X. Ma, J. Li, L. Hai and Y. Wu, Tetrahedron, 2015, 71, 6928 CrossRef CAS.
- (a) Y. Jia, D. Katayev and E. P. Kündig, Chem. Commun., 2010, 46, 130 RSC; (b) J. X. Hu, H. Wu, C. Y. Li, W. J. Sheng, Y. X. Jia and J. R. Gao, Chem.–Eur. J., 2011, 17, 5234 CrossRef CAS PubMed; (c) D. Soló, F. Mariani, I. Fernández and M. A. Siera, J. Org. Chem., 2012, 77, 10272 CrossRef PubMed; (d) D. Soló, F. Mariani and I. Fernández, Adv. Synth. Catal., 2014, 356, 3237 CrossRef; (e) L. Yin, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7620 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c5ra23837d |
| This journal is © The Royal Society of Chemistry 2015 |