
Morphology adjustment of SnO2 and SnO2/CeO2 one dimensional nanostructures towards applications in gas sensing and CO oxidation
Yunshi Liua,
Ping Yang*a,
Jia Lia,
Katarzyna Matras-Postolekb,
Yunlong Yuea and
Baibiao Huangc
aSchool of Material Science and Engineering, University of Jinan, Jinan, 250022, P. R. China. E-mail: mse_yangp@ujn.edu.cn; Fax: +86 531 87974453; Tel: +86 531 89736225
bFaculty of Chemical Engineering and Technology, Cracow University of Technology, Krakow, 31-155, Poland
cState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, P. R. China
First published on 13th November 2015
Abstract
SnO2 and SnO2/CeO2 one dimensional (1D) nanostructures with various morphologies including solid nanofibers (NFs), nanobelts (NBs), nanotubes (NTs), and wire-in-tubes (WTs) were successfully prepared via a single-spinneret electrospinning process and a subsequent heat-treatment by adjusting the heating rate and the amount of CeO2. The interesting morphology evolution of samples from NTs to NBs was reported with increasing amounts of CeO2. In addition, SnO2 particles existed on the surface of the CeO2 matrix to form diverse structures in the 1D nanofibers. The band gap of the composite oxides decreased with the addition of CeO2. Compared with other nanostructures, SnO2/CeO2 NTs exhibited superior gas sensing properties, such as the highest response to ethanol. Due to the existence of Ce3+ and oxygen vacancies and the hollow structure, SnO2/CeO2 NTs revealed great CO oxidation performance, indicating the enhanced interactions between the catalyst and the target gas. These excellent properties were attributed to the prominent 1D hollow morphology, the good dispersion of SnO2 nanoparticles on the surface of the CeO2 matrix, the ideal crystallinity, and the composite interactions between the two oxides. In addition, the current method could be utilizable to fabricate other metal oxides with various morphologies for property controlling and important applications.
Introduction
As a special morphology, one-dimensional (1D) nanostructures, including solid nanofibers (NFs), nanobelts (NBs), nanotubes (NTs), and wire-in-tubes (WTs), are of great interest in a wealth of applications, such as energy conversion,1 tissue engineering scaffolds,2 gas sensing,3 catalysis,4 and so on. To date, a variety of methods have been invented to fabricate 1D nanostructures, for example, template-assisted synthesis,5 solvothermal route,6 organic chemical vapor deposition,7 structure-selective synthesis.8 Notably, electrospinning is a simple, versatile, and cost-effective technique for preparing 1D nanostructures with controllable compositions, diameters, and morphologies for a wealth of applications. This strategy links the dimension and morphology of nano- and macroscale materials to properties in a flexible manner. Many methods including polymer template method,9 coaxial electrospinning technique,10 and phase separation co-electrospinning process,11 have been reported for fabrication of 1D NTs. However, these procedures often suffer from tedious processes, templates and polymers. In addition, to create other 1D morphologies, we have to find other more effective method.With the development of science and technology, scientists pay much more attention to the environmental problems and sustainable development, such as gas sensing and CO oxidation. Ethanol sensor is one of the most popular gas sensors due to broad applications in medical process, breathalyzer, and food industries. The studies of the catalytic properties of CO oxidation using 1D oxide catalysts are very important in chemical engineering, such as air purification and automotive exhaust cleaning field. Besides a reasonable structure, a suitable composition of the functional materials is also very important. Among those 1D nanocomposites, semiconductor-based complex oxides, such as ZnO/SnO2,12 TiO2/CuO,13 In2O3/CeO2,14 and ZnO/TiO2,15 have attracted extensive attentions. SnO2, as an excellent n-type IV–VI semiconductor, with a wide band gap of ∼3.6 eV, has been proved to be an important gas sensing material with applications in detecting both reducing and oxidizing gases. As a well-known rare earth oxide, CeO2 exhibits promising properties in catalytic and gas sensing area due to its particular characteristics arising from the 4f electronic shells, rich oxygen vacancies and low redox potential between Ce3+ and Ce4+. It is expected that the composition of CeO2 and SnO2 could remarkably enhance the performance due to the strong interactions between two individual components.
Best to our knowledge, little attention has been paid to the control of the morphology of nanostructures fabricated by single-spinneret electrospinning. In this paper, for the first time, we observed a morphology evolution of SnO2/CeO2 composites from NTs to NBs. The formation mechanism of the nanostructures was studied. In addition, their bifunctional gas sensing properties and CO oxidation performance based on different mechanisms are also systematically studied. The fibers with various morphologies display different ethanol sensing properties and catalytic performances due to the special feature of the structure and the oxygen vacancies.
Experimental
Materials
N,N-Dimethylformamide (DMF) was taken from Tianjin Chemical Reagent Institute. Poly(vinylpyrrolidone) (PVP; Mw ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) was purchased from Aladdin Reagent Company. Ce(NO3)3·6H2O and SnCl2·2H2O were purchased from Sinopharm Chemical Reagent Company. All chemicals were analytical grade reagents and they were used directly without any further purification.
300000) was purchased from Aladdin Reagent Company. Ce(NO3)3·6H2O and SnCl2·2H2O were purchased from Sinopharm Chemical Reagent Company. All chemicals were analytical grade reagents and they were used directly without any further purification.
Preparation of 1D SnO2 and SnO2/CeO2 nanostructures
In a typical synthesis, different amount of SnCl2·2H2O and Ce(NO3)3·6H2O were added to 12 mL of DMF with vigorous stirring for 1 h. Then, 2 g of PVP was added to the above solution and the mixtures were allowed to stir for 24 h. After that, viscous mixed solutions of inorganic salts and PVP were obtained. The detailed preparation conditions and properties of the samples were listed in Table 1.| Sample | Molar ratioa | Heating rate (°C min−1) | Morphology | Average diameter (nm) | Band gap (eV) |
|---|---|---|---|---|---|
| a Sn/Ce molar ratio in precursors. | |||||
| Sn-NFs | N/A | 1 | Nanofibers | 170 | 3.74 |
| Sn-NTs-1 | N/A | 5 | Nanotubes | 600 | 3.73 |
| Sn-NTs-2 | N/A | 5 | Nanotubes | 330 | 3.76 |
| Sn–Ce-WTs | 20:1 |
5 | Wire-in-tubes | 200 | 3.53 |
| Sn–Ce-NTs | 10:1 |
5 | Nanotubes | 200 | 3.3 |
| Sn–Ce-NFs | 5:1 |
5 | Nanofibers | 225 | 3.07 |
| Sn–Ce-NBs | 1:1 |
5 | Nanobelts | 500 | 2.91 |
The as-spun fibers were prepared via a typical single-spinneret electrospinning method.16 Each solution was loaded in a 5 mL syringe fitted with a stainless steel needle with ∼0.8 mm inner diameter. The syringe was fixed horizontally on the pump and the optimal feed rate was chosen at 7 μL min−1. An electrospinning process was performed by applying a DC voltage of 15 kV to the needle and the tip-to-collector was 15 cm. As the solvent evaporated, the as-spun fibers were obtained on the collector. After that, these samples were calcined at 550 °C for 3 h with different heating rates. After the fibers were naturally cooled down to room temperature, the target samples were obtained.
Characterization
The morphology observation of samples was carried out using a field-emission scanning electron microscopy (FESEM, QUANTA 250 FEG, FEI, America). The transmission electron microscopy (TEM) images were recorded on a JEM-2010 transmission electron microscope and a transmission/scanning transmission electron microscope (TEM/STEM, Tecnai F20, FEI). The average diameter of samples was calculated based on randomly selected fibers. The crystal structures and phase composition of samples were observed using an X-ray diffraction (XRD) meter (Bruker D8-Advance, Germany). X-ray photoelectron spectroscopy (XPS) experiments were investigated using an X-ray photoelectron spectroscopy (XPS, ESCALAB 250) with a monochromatic Al KR source. All the binding energies were referenced to the C 1s peak at 284.7 eV of the surface adventitious carbon. The fitted peaks in the XPS spectra were analyzed using the Avantage 5.52 software. The UV-vis absorption spectra were measured on a Hitachi U-4100 spectrophotometer. The band gaps of samples were estimated from the adsorption spectra as listed in Table 1.Gas sensing measurement
Gas sensing performance was evaluated using a WS-30A system (Weisheng Instruments Co., Zhengzhou, China). The as-prepared sample was mixed with ethanol to form a paste. Then, the paste was coated on a ceramic tube, on which a pair of gold electrodes had been previously printed. A Cr–Ni heating wire was inserted into the ceramic tube as a resistor. Before measurement, the sensors were dried and aged for about 5 days. When testing, the saturated target gas was injected into the test chamber (about 1 L) by a disposable syringe. After fully mixed with ambient air, the sensor was put into the chamber. The sensor was taken out to recover in ambient air after the response reached a constant value. The gas sensor resistance and response values were obtained by the analysis system automatically. In the experimental process, a working voltage of 5 V was applied on the sensor. The response of the sensor in air or in a target gas could be measured by monitoring the voltage across the reference resistor. The gas sensor response value was defined as below:| Response = Rgas/Rair | (1) |
CO catalytic oxidation
CO oxidation properties were measured on a fixed-bed flow reactor with 50 mg of samples under atmospheric pressure. The reaction vessel was a quartz tube with an inside diameter of 6 mm. A gas mixture with a total flow rate of 50 mL min−1 flowed through the quartz tube and the feed gas was 1% CO, 10% O2, and 89% N2 in volume ratio. The programmed-temperature rate was 2 °C min−1 which was controlled in an electrical furnace. A thermocouple placed in the middle of the reactor was used to monitor the reaction temperature. The catalyst was directly exposed to reaction gas without any pretreatment. The composition analysis of the effluent gas was carried out by a gas analyzer. The CO conversion ratio was observed on the basis of the CO consumption and CO2 formation.Results and discussion
Fig. 1a to c show the typical SEM images of samples Sn-NFs, Sn-NTs-1, and Sn-NTs-2. These randomly oriented 1D nanostructures were all composed of lots of uniform nanoparticles. The image in Fig. 1a indicates that sample Sn-NFs revealed a solid fibrous morphology with a diameter of ∼170 nm due to the low heating rate during calcination process. As increasing the heating rate to 5 °C min−1, we obtained well-defined SnO2 NTs with a diameter of 600 nm, as shown in Fig. 1b. The wall of the tube is composed of a layer of SnO2 nanoparticles, suggesting the wall thickness is ∼70 nm. As increasing the amount of SnCl2 in spinning precursor solution, SnO2 tubes with thick walls are obtained. The inset in Fig. 1d depicts the TEM image of sample Sn-NTs-1, clearly indicating a large quantity of tube-like structures composed of numerous SnO2 nanoparticles. More detailed information of the SnO2 NTs and the crystal structures can be observed by high-magnification TEM, as shown in Fig. 1d. The lattice fringes with a spacing of 0.33 nm can be observed, which is in agreement with the spacing of (110) planes of SnO2. Furthermore, the clear crystal lattice shows sample Sn-NTs-1 with good crystallinity. The various morphologies indicate that both of the content in spinning solution and the heating rate during the calcination process plays an important role in preparing 1D structures via electrospinning. | ||
| Fig. 1 (a, b, and c) SEM images of (a) sample Sn-NFs, (b) sample Sn-NTs-1, and (c) sample Sn-NTs-2. (d) HRTEM image of sample Sn-NTs-1. Inset in (d) shows a TEM image with low magnification. | ||
To investigate the effect of Ce amounts on the morphologies of SnO2/CeO2 composite structures, SEM images are given in Fig. 2. As we added the CeO2 component, we observed the interesting morphology evolution of the samples from NTs to NBs. The diameters of them are ∼500, ∼225, ∼200, and ∼200 nm for sample Sn–Ce-NBs, sample Sn–Ce-NFs, sample Sn–Ce-NTs, and sample Sn–Ce-WTs, respectively. It can be observed that all the 1D structures are long, continuous, and randomly oriented. When the molar ratio of Sn and Ce is 1:1, NBs with uniform nanoparticles on their surface are observed. SnO2/CeO2 NFs, NTs, and WTs were prepared with the further addition of Ce(NO3)3 in the spinning solution, as shown in Fig. 2b, c, and d, respectively. As can be investigated from the funny morphology evolution, the hollow structures gradually decrease and the morphologies of the samples are more likely to form ribbon-like structure with the increase of Ce amount. Therefore, the morphologies of SnO2/CeO2 samples are greatly influenced by the amounts of CeO2. These results are consistent with our previous report.16 However, we also found that the particles with size ranging from 30 to 70 nm were exposed on the surface of 1D structures. Compared with the CeO2 1D structures that we prepared before, it was found that the outer particles on the surface of composite structures are SnO2 particles and the inner component is CeO2. In other words, the SnO2 component is more likely to move to the surface of the fibers during calcination. As can be seen from the samples, the SnO2 particles always exist on the surface of the CeO2 matrix to form various structures.
 | ||
| Fig. 2 SEM images of (a) sample Sn–Ce-NBs, (b) sample Sn–Ce-NFs, (c) sample Sn–Ce-NTs, and (d) sample Sn–Ce-WTs. The insets in (a) and (b) show the images with high magnification. | ||
XRD measurement was purposely carried out to provide further insight into the crystallinity of the samples. All samples are well-crystallized and impurity peaks are not observed in Fig. 3, indicating the high purity of the products. As can be seen, the prominent peaks correspond to (110), (101), (211) crystal lattice planes and all other smaller peaks coincide with the corresponding peaks of the tetragonal rutile structure of SnO2 (JCPDS no. 41-1445). As the addition of Ce, the samples display some additional unconspicuous diffraction peaks which can be correspond to CeO2 (JCPDS no. 65-2975), as marked by stars. As can be seen from the diffraction patterns of sample Sn–Ce-WTs, the CeO2 peaks are weaker than those of SnO2, in accordance with the presence of small quantities of CeO2. When the amounts of Ce is increasing, the diffraction peaks which belong to CeO2 become more and more apparent, indicating the formation of the pure cubic CeO2 phase. For SnO2/CeO2 samples, there are no obvious peak shifts or any trace of other phases besides rutile SnO2 and cubic CeO2, indicating the well-crystallized composite structures.
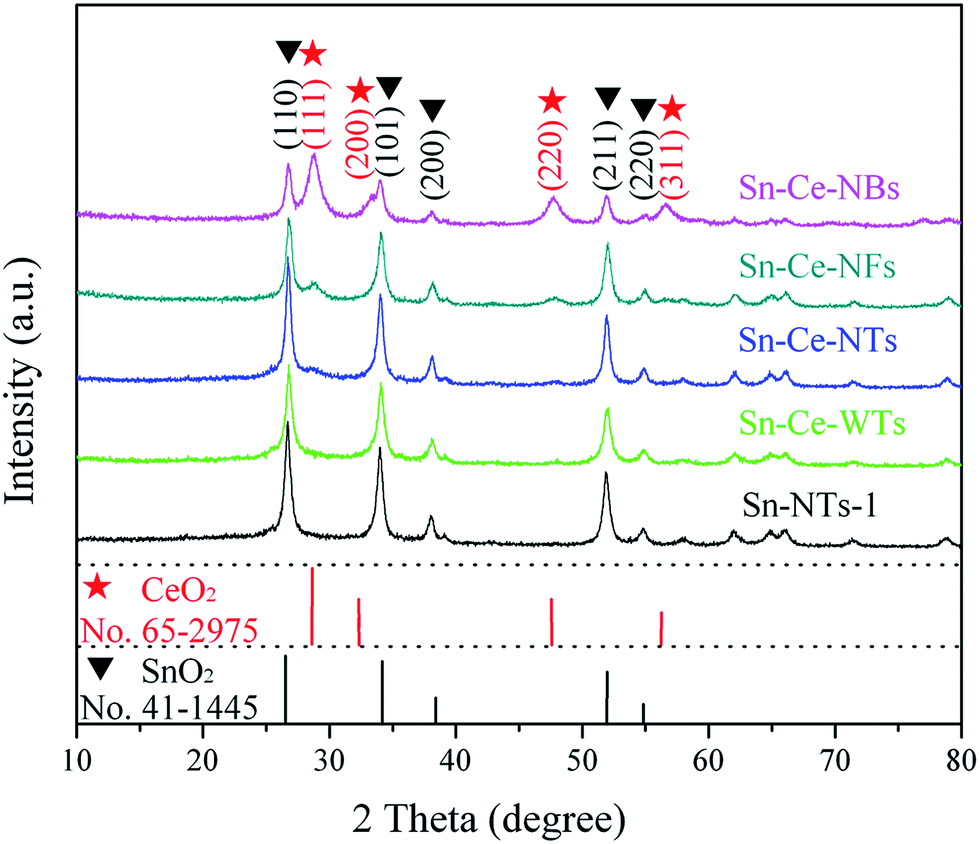 | ||
| Fig. 3 XRD patterns of SnO2 and SnO2/CeO2 composite samples. | ||
In order to determine the optical band gap and associated properties, the optical absorbance measurements were carried out at room temperature using an ultraviolet-visible diffused reflectance spectrometer. Furthermore, the band gap energies of the samples are investigated from a plot of (αhν)2 vs. energy (hν), as shown in Fig. 4. For a crystalline semiconductor, the optical absorption near the band edge follows eqn (2)17,18
| αhν = A(hν − Eg)n/2 | (2) |
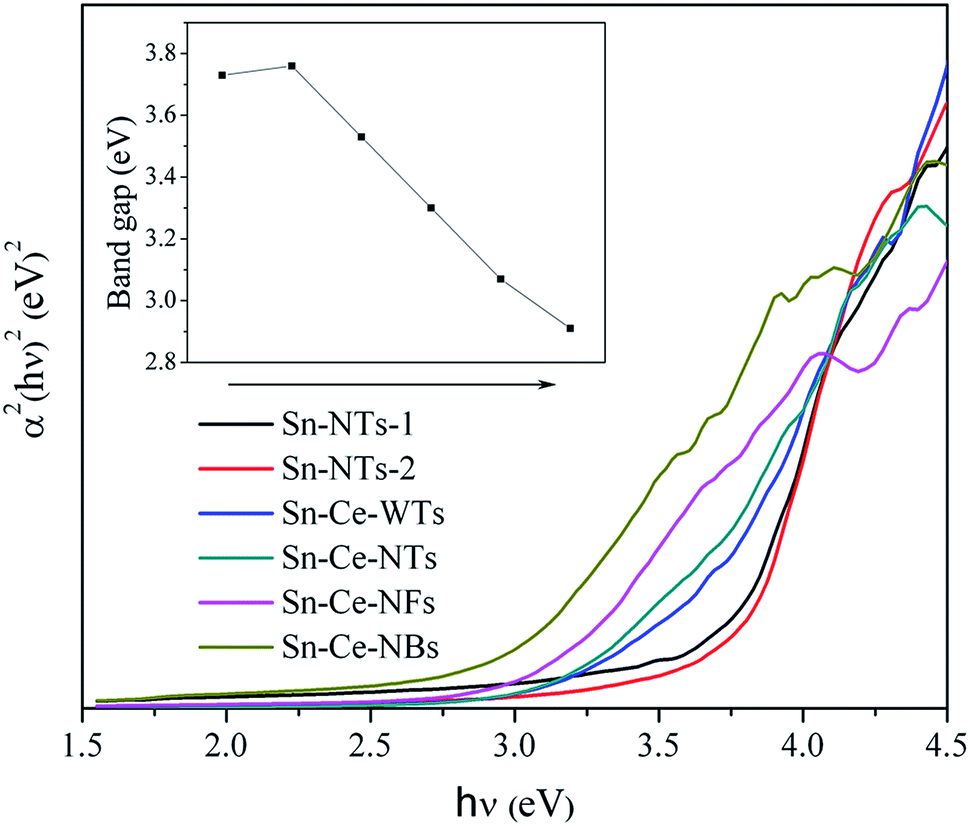 | ||
| Fig. 4 Plots of (αhν)2 vs. energy (hν) for the band gap energies. Inset is the corresponding band gaps of each sample. | ||
Based on the above, in order to further confirm the effect of the SnO2 structure and CeO2 distribution, sample Sn–Ce-NBs and Sn–Ce-NTs are proposed and the details are shown in Fig. 5. The inset of each image shows the TEM image, indicating the typical morphologies of NBs and NTs, which is in agreement with the SEM images. It could be easily confirmed that both of SnO2/CeO2 NBs and NTs were highly crystalline in the HRTEM images. The interplanar spacing of the outer particles of the samples was measured to be 0.237 and 0.334 nm, which correspond to the d spacing of the (200) and (110) planes of the rutile structure of SnO2, respectively. In addition, the interplanar spacing of the inner particles of the samples was measured to be 0.270 and 0.312 nm, which correspond to the d spacing of the (200) and (111) planes of the cubic structure of CeO2. It is also easy to find that the SnO2 and CeO2 grains are connected to each other by grain boundaries, suggesting the good crystallinity of the samples. As is depicted, the obvious distribution of outer SnO2 grains and inner CeO2 grains is consistent with our previous idea in the analysis of SEM images, indicating the significant effect of the Ce amounts on the morphology. Therefore, the SnO2 particles always exist on the surface of the CeO2 matrix to form various structures. These special structures are similar to core–shell structures, which may contribute significantly to the electrical conduction and gas-sensing properties of the material.
 | ||
| Fig. 5 HRTEM images of (a) sample Sn–Ce-NBs and (b) sample Sn–Ce-NTs. Inset shows the corresponding TEM images of each sample. | ||
On the basis of the experimental observations, a possible formation mechanism of SnO2 nanostructures and SnO2/CeO2 composite nanostructures is proposed and the overall morphology evolution of the nanostructures is displayed in Scheme 1. First of all, due to the high voltage applied on the precursor solution, PVP molecular chains were positively charged during the electrospinning process. Under appropriate conditions, the liquid jets of polymer solution emitted and the as-spun fibers were collected on the aluminum foil. Then various nanostructures were obtained after calcination process. For SnO2 nanostructures, we successfully prepared SnO2 solid fibers with a heating rate of 1 °C min−1. PVP were slowly decomposed at this slow heating rate and the gases were released slowly during the calcination process, resulting in the formation of solid structure. As increasing the heating rate, the decomposition of PVP became faster. In addition, the release rate of gases inside the fiber is larger than the diffusion rate of gases through the fiber surface, leading to the increase of the pressure inside of the fiber. Then hollow structures were formed.22 With increasing the amount of SnCl2 in spinning precursor solution, the results suggested that the products were remained with tubular structure with thicker walls. Therefore, for pure SnO2, fast heating rates are required to facilitate hollow fiber formation.
 | ||
| Scheme 1 Proposed mechanism of the formation of SnO2 nanostructures and SnO2/CeO2 composite nanostructures prepared via single-spinneret electrospinning. | ||
Various SnO2/CeO2 nanostructures were obtained via this single-spinneret electrospinning method. As increasing the amount of Ce, we found an interesting morphology evolution from NTs to NBs. For sample Sn–Ce-WTs and Sn–Ce NTs, we observed wire-in-tube and tubular structure with a high heating rate due to the addition of effect of gas expansion to the Kirkendall effect.23 Then, SnO2/CeO2 NFs and NBs were prepared via adjusting the amounts of Ce during electrospinning. Hence, the amounts of Ce played a more important role in adjusting morphology of SnO2/CeO2 composite structures. In addition, SnO2 particles were more likely to form on the surface of nanostructures regardless of the morphologies, as can be previously proved by SEM and TEM images. As is known in calcination process, the mechanism is Kirkendall effect, where the diffusion of precursor toward the surface and away from the center of the fibers due to the concentration gradient. This results in the closer formation of particles to the fiber surface. After decomposition of PVP, larger particles remain self-supported on the exterior, forming the shell of the fiber, whereas particles below 30 nm in size collapse to the center of the fiber.24 During this diffusion process, the SnO2 particles remained on the exterior of the fiber due to their larger size than that of CeO2 particles. Because of this possible mechanism, these special structures prepared via a single-spinneret electrospinning method are similar to core–shell structures, which may be used as a model to fabricate more other metal oxides with various morphologies.
In Fig. 6, to further investigate the chemical state and surface composition of the elements, XPS spectra of sample Sn–Ce-NTs were studied. The survey spectra of the sample confirm the presence of Sn, O, Ce, and C, as shown in Fig. 6a. In Fig. 6b, due to the asymmetry of the O 1s profile of sample Sn–Ce-NTs, the peak could be dissolved into three symmetrical peaks. The three peaks are locating at 528.8, 530.1, and 531.5 eV, respectively, suggesting three different kinds of O species in the sample. The peak at 531.5 eV corresponds to chemisorbed oxygen or hydroxyl ions such as OH−, O−, and O2− at the surface of SnO2.25–27 The peaks at 528.5 and 530.1 eV are assigned to core levels of oxygen coordinated to Sn2+ and Sn4+ in the O 1s, respectively. As previous work reported, the O 1s core level of Ce2O3 showed a higher binding energy than that of CeO2.28 It is difficult to clearly distinguish the O 1s peaks of Sn2+, Sn4+, Ce3+, and Ce4+ due to close peak position. However, after introducing Ce to the sample, the O 1s peaks are gradually broadened at low energy, indicating the decrease of the binding energy of the oxygen and the formation of surface oxygen vacancies.
 | ||
| Fig. 6 (a) Survey, (b) O 1s, (c) Sn 3d, and (d) Ce 3d high resolution XPS spectra of sample Sn–Ce-NTs. | ||
Fig. 6c shows characteristic spin–orbit for Sn 3d5/2 and Sn 3d3/2 of sample Sn–Ce-NTs. The Sn 3d5/2 can be resolved into two peaks, locating at 486.2 and 486.9 eV, respectively. The former peak is assigned to Sn2+, coming from SnO2, while the latter peak corresponds to Sn4+, resulting from SnO2. Due to the addition of Ce, the formation of Sn2+ may increase the surface deficiency of SnO2 crystallites, resulting in active sites for reactions. The Ce 3d XPS spectrum of sample Sn–Ce-NTs is shown in Fig. 6d. It seems more complex due to the multiplicity of final states reached during the Ce 3d photoionization process.29,30 Based on the previous reported work, the Ce 3d level can be dissolved into two series of peaks.31,32 The peaks labeled u0, u, u1, u2, and u3 are the result of the 3d3/2 spin–orbit states, and those labeled v0, v, v1, v2, and v3 are assigned to the corresponding 3d5/2 states. Among these peaks, six typical peaks marked by u, u2, u3, v, v2, and v3 are the characteristic peaks of Ce4+, and the rest of them labeled as u0, u1, v0, and v1 are in agreement with the previous report about Ce3+. In other words, sample Sn–Ce-NTs has a mixture of Ce3+/Ce4+ oxidation states, indicating that the large amount of oxygen vacancies exist on the surface of the sample.
It is significant to investigate the influence of different morphologies, special structure, and large amount of oxygen vacancies on their application performance. The response of SnO2/CeO2 nanostructures gas sensors to 100 ppm ethanol as a function of operating temperature is shown in Fig. 7a. As is known to all, the operating temperature has a significant effect on the response of a semiconductor base gas sensor. All the sensors have the same changing trend that the responses first increase with temperature, up to 320 °C, and then gradually decrease. It can be seen that the optimum working temperature for ethanol gas detection is about 320 °C and sample Sn–Ce-NTs gas sensor has the largest response (∼49.1) at this temperature, which is four times higher than that of pure SnO2 NTs gas sensor. Also, the result is better than the response (∼31) of SnO2/CeO2 sensor in previous report.33 This is ascribed to the addition of Ce component to the composite, providing more absorption site and vacancies in the gas sensing test. It is noted that the responses of all SnO2/CeO2 composite nanostructures gas sensors to ethanol have remarkable improvement except sample Sn–Ce-NBs. The reasonable explanation is that the ribbon-like structure cannot provide large amount of active sites like hollow structure. In addition, the introducing of excessive Ce has some negative effect on the response to ethanol, resulting in the decrease of response of sample Sn–Ce-NBs.
 | ||
| Fig. 7 (a) Relationships of the operating temperature versus the response of the samples based sensor toward 100 ppm ethanol, respectively; (b) response and recovery characteristic curves of SnO2 nanostructures based sensor to 50–1000 ppm ethanol, respectively; (c) the cross-response of the sensors to 100 ppm ethanol, ammonia, benzene, toluene, chloroform, and hexane. (d) Response and recovery characteristic curves of SnO2/CeO2 composite nanostructures based sensor to 50–1000 ppm ethanol, respectively. | ||
To further investigate the gas sensing behavior, the dynamic response of pure SnO2 nanostructures and SnO2/CeO2 composite nanostructures are observed. As shown in Fig. 7b, the response of sample Sn-NTs-1 is higher than that of sample Sn-NTs-2, indicating the effect of wall thickness of NTs on gas sensing behavior. Fig. 7d shows that sample Sn–Ce-NTs exhibits the excellent response to ethanol among these samples because of the hollow structure and oxygen vacancies, which are consistent with our previous analysis. Based on the dynamics, the response and recovery time of different samples were obtained and compared. As shown in Table 2, the response of sample Sn–Ce-WTs to 100 ppm ethanol is about 33.8, which is similar to that of sample Sn–Ce-NFs (35.4). This phenomenon is caused by the solid structure, resulting in the less active sites and the decrease of contact area for target gas.
The sensing mechanism for SnO2 can be mainly attributed to the adsorption and desorption of the target gas molecules on the surface of the samples. This process would cause the change in resistance in air and in target gas. It is worth mentioning that the adsorption mechanism was greatly influenced by the oxygen vacancies on the surface. Compared with pure SnO2 samples, SnO2/CeO2 composite samples exhibited enhanced ethanol sensitivity, due to the additional redox couple of Ce3+ and Ce4+ as proved by XPS. However, because of the relatively poor conductivity, the excessive addition of Ce could make more CeO2 nanoparticles formed in composite nanostructures which hinder the electron transfer in gas sensing process. Thus, the relatively high gas sensing property of sample Sn–Ce-NTs could be attributed to the appropriate introduction of Ce and the hollow structure.
CO catalytic oxidation was carried out to investigate the catalytic performance of pure SnO2 NTs and SnO2/CeO2 composite nanostructures, as shown in Fig. 8. The details are shown in Table 2. Due to the continuous oxidation of CO, the CO conversion ratios were gradually increased with temperature. The CO catalytic properties of all the SnO2/CeO2 composite nanostructures are better than that of pure SnO2 NTs, indicating that the introducing of Ce has positive effect on the catalytic behavior. Especially, the onset oxidation temperature (T10) for Sn–Ce-NTs is 203 °C, while the complete CO oxidation temperature (T100) is 317 °C. Both of the two temperatures are remarkably below the rest samples, indicating the excellent catalytic performance for CO oxidation. It is well known, both of SnO2 and CeO2 can promote the CO oxidation reaction due to their large number of oxygen vacancies form the existence of Sn2+/Sn4+ and Ce3+/Ce4+. In addition, the structure of the sample also played an important role in catalysis. The CO oxidation properties of sample Sn–Ce-NTs and Sn–Ce-WTs are better than that of sample Sn–Ce-NFs with a solid structure, suggesting that the hollow structure may provide more active sites and contact area during catalysis. However, sample Sn–Ce-NBs exhibited low completed CO oxidation temperature compared with sample Sn–Ce-WTs. This is ascribed to the different amounts of Ce in composite samples. Therefore, the introduction of Ce to the samples could significantly enhance the catalytic performance for CO oxidation. Furthermore, it should be mentioned that both the ratios of Ce and the structure of samples could influence the CO oxidation properties of SnO2/CeO2 composites.
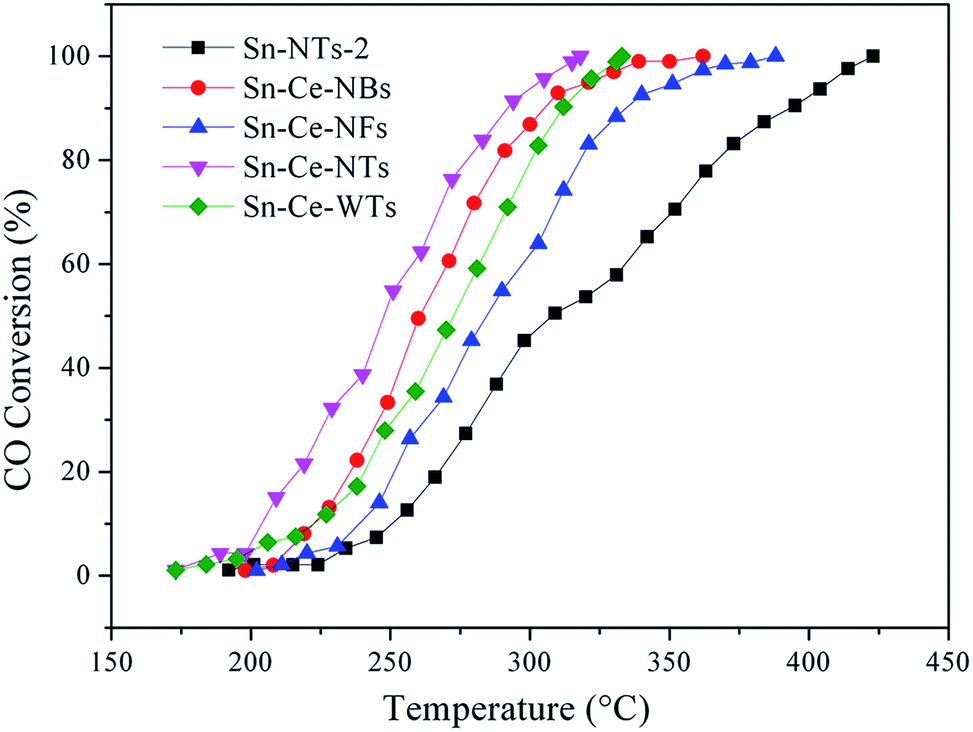 | ||
| Fig. 8 CO oxidation of samples Sn-NTs-2, Sn–Ce-NBs, Sn–Ce-NFs, Sn–Ce-NTs, and Sn–Ce-WTs. | ||
Conclusions
1D SnO2 and SnO2/CeO2 fibers with diverse morphologies were fabricated via a single-spinneret electrospinning by adjusting the heating rate and the amounts of Ce. Their applications in gas sensing and CO oxidation were investigated. It can be concluded that the nanostructures were uniformly composed of nanoparticles and as increasing the amounts of Ce, the morphology evolution from NTs to NBs was observed. The band gap of the composite oxides decreases with the addition of CeO2. The XPS analysis showed the existence of Sn2+ and Ce3+, indicating the improvement of surface oxygen vacancies. Sample Sn–Ce-NTs had the high response to ethanol and excellent catalytic performance for CO oxidation, which can be attributed to the hollow structure, good crystallinity, and the interactions between SnO2 and CeO2. The present work suggests that many other functional metal oxides with diverse morphologies could be prepared via this single-spinneret electrospinning and their potential applications can benefit from their special structures.Acknowledgements
This work was supported in part by the project from National Basic Research Program of China (973 Program, 2013CB632401), the program for Taishan Scholars, the projects from National Natural Science Foundation of China (51202090, 51302106, 51402123, and 51402124).Notes and references
- K. Peng and S. Lee, Adv. Mater., 2011, 23, 198 CrossRef CAS PubMed.
- D. Kai, M. Prabhakaran, G. Jin and S. Ramakrishna, J. Biomed. Mater. Res., Part B, 2011, 98, 379 CrossRef PubMed.
- S. Bai, S. Chen, Y. Zhao, T. Guo, R. Luo, D. Li and A. Chen, J. Mater. Chem. A, 2014, 2, 16697 CAS.
- S. Xu, D. Sun, H. Liu, X. Wang and X. Yan, Catal. Commun., 2011, 12, 514 CrossRef CAS.
- G. Zhang and X. Lou, Sci. Rep., 2013, 3, 1470 Search PubMed.
- W. Huang, Y. Jiang, X. Li, X. Li, J. Wang, Q. Wu and X. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8845 CAS.
- Z. Fan, J. Yan, T. Wei, G. Ning, L. Zhi, J. Liu, J. Liu, D. Cao, G. Wang and F. Wei, ACS Nano, 2011, 5, 2787 CrossRef CAS PubMed.
- J. Wang, J. Zhang, B. Asoo and G. Stucky, J. Am. Chem. Soc., 2003, 125, 13966 CrossRef CAS PubMed.
- R. Caruso, J. Schattka and A. Greiner, Adv. Mater., 2001, 13, 1577 CrossRef CAS.
- D. Li, J. McCann, Y. Xia and M. Marquez, J. Am. Ceram. Soc., 2006, 89, 1861 CrossRef CAS.
- R. Ab Kadir, Z. Li, A. Sadek, R. Abdul Rani, A. Zoolfakar, M. Field, J. Ou, A. Chrimes and K. Kalantar-zadeh, J. Phys. Chem. C, 2014, 118, 3129 CAS.
- L. Xu, R. Xing, J. Song, W. Xu and H. Song, J. Phys. Chem. C, 2013, 1, 2174 CAS.
- S. Lee, H. Bai, Z. Liu and D. Sun, Water Res., 2013, 47, 4059 CrossRef CAS PubMed.
- L. Xu, H. Song, B. Dong, Y. Wang, J. Chen and X. Bai, Inorg. Chem., 2010, 49, 10590 CrossRef CAS PubMed.
- S. Hwang, J. Song, Y. Jung, O. Kweon, H. Song and J. Jang, Chem. Commun., 2011, 47, 9164 RSC.
- Y. Liu, H. Chen, J. Li and P. Yang, RSC Adv., 2015, 5, 37585 RSC.
- M. Butler, J. Appl. Phys., 1977, 48, 1914 CrossRef CAS.
- J. Zeng, H. Wang, Y. Zhang, M. Zhu and H. Yan, J. Phys. Chem. C, 2007, 111, 11879 CAS.
- L. Shi and H. Lin, Langmuir, 2011, 27, 3977 CrossRef CAS PubMed.
- L. Xu, B. Dong, Y. Wang, X. Bai, J. Chen, Q. Liu and H. Song, J. Phys. Chem. C, 2010, 114, 9089 CAS.
- T. Ma, Z. Yuan and J. Cao, Eur. J. Inorg. Chem., 2010, 5, 716 CrossRef.
- Y. Zhang, J. Yang, Q. Li and X. Cao, J. Cryst. Growth, 2007, 308, 180 CrossRef CAS.
- J. Wu, D. Zeng, X. Wang, L. Zeng, Q. Huang, G. Tang and C. Xie, Langmuir, 2014, 30, 11183 CrossRef CAS PubMed.
- S. Cavaliere, S. Subianto, I. Savych, M. Tillard, D. Jones and J. Roziere, J. Phys. Chem. C, 2013, 117, 18298 CAS.
- Y. Nagasawa, T. Choso, T. Karasuda, S. Shimomura, F. Ouyang, K. Tabata and Y. Yamaguchi, Surf. Sci., 1999, 433, 226 CrossRef.
- A. Kolmakov, S. Potluri, A. Barinov, T. Mentes, L. Gregoratti, M. Niño, A. Locatelli and M. Kiskinova, ACS Nano, 2008, 2, 1993 CrossRef CAS PubMed.
- D. Yang, I. Kamienchick, D. Youn, A. Rothschild and I. D. Kim, Adv. Funct. Mater., 2010, 20, 4258 CrossRef CAS.
- G. Praline, B. E. Koel, R. L. Hance, H. I. Lee and J. M. White, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 17 CrossRef CAS.
- C. Ho, J. Yu, T. Kwong, A. C. Mak and S. Lai, Chem. Mater., 2005, 17, 4514 CrossRef CAS.
- A. Pfau and K. D. Schierbaum, Surf. Sci., 1994, 321, 71 CrossRef CAS.
- B. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 5162 CrossRef CAS.
- C. Force, E. Roman, J. Guil and J. Sanz, Langmuir, 2007, 23, 4569 CrossRef CAS PubMed.
- W. Qin, L. Xu, J. Song, R. Xing and H. Song, Sens. Actuators, B, 2013, 185, 231 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |