
Electron competitive migration regulating for dual maxima of water photolysis†
Xianqun Chenab,
Liping Lia,
Yangsen Xua,
Yuelan Zhanga and
Guangshe Li*a
aKey Laboratory of Design and Assembly of Functional Nanostructures, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: guangshe@fjirsm.ac.cn; Fax: +86-591-63179426; Tel: +86-591-63179426
bCollege of Materials Science and Engineering, Fujian Normal University, Fuzhou 350007, P. R. China
First published on 15th December 2015
Abstract
Electron competitive migration between the conduction band and charge trap centre is the key in governing the catalytic activity and the relevant applications of semiconductor nanomaterials, which is however poorly understood yet. Herein, we systematically studied the electron competitive migration in defective SnO2 nanoparticles through hybridizing with a polymer electron donor, graphitic carbon nitride (g-C3N4). When varying the mass ratio of defective SnO2 from 5% to 70%, an increase of surface-charge trapping centres (oxygen vacancies) in SnO2 effectively regulated the electron competitive migration. As a consequence, dual catalytic activity maxima were observed in hydrogen generation from water splitting under visible light irradiation (λ > 420 nm). For instance, the relative mass ratios at 10% and 40% yielded maximum hydrogen generation rates of 54.3 μmol h−1 g−1 and 44.3 μmol h−1 g−1, respectively, far beyond that of 27.9 μmol h−1 g−1 for pure g-C3N4. Strikingly, the photon–hydrogen conversion efficiency also showed dual maxima values as SnO2 mass ratio changes. These abnormal observations were comparatively investigated via XPS, EPR and photoluminescence spectra in solid state and aqueous environments. It is demonstrated that electron competitive migration was primarily caused by oxygen vacancies on the SnO2 surface, which plays a key role in creating the dual catalytic activity maxima in water splitting.
1 Introduction
Hydrogen energy gained from water splitting over supported semiconductors under sunlight has attracted world-wide attention. The conversion efficiency directly determines whether this conversion scheme can be applied to solving the energy shortage crisis. Much effort has been taken to develop photocatalysts with an aim to solve the fundamental problems of energy conversion efficiency from photons to hydrogen since the first report by Fujishima and Honda.1 Since then, multiple modification of photocatalysts has become an important method to improve the photon utilization, which includes (i) improving light response range through doping or introducing bulk defect state/surface disorder, such as nitrogen-doped TiO2,2 and disorder-engineered black TiO2,3 (ii) introducing surface defects as charge trap centres to enhance the charge separation rate. For instance, Tang and coworker4 have introduced oxygen vacancies in ZnO which enhances the photocatalytic activity; and (iii) semiconductor-based hybrids via hybridizing different semiconductors to enhance the photon–hydrogen conversion efficiency. For example, hybridizing graphitic carbon nitride (g-C3N4) with metal oxides or sulfides, like ZnO,5–7 CdS,8–10 TiO2,11–15 WO3,16,17 Bi2WO6,18 and other kind of hybrids have been investigated widely.19–21 In this regard, photo-generated carrier's migration has shown impacts on surface photocatalytic redox reaction, and photocatalytic ability could be thus enhanced by increasing the separation rate of photo-generated carriers. Though the methods mentioned above have given rise to the enhanced light response ability, introducing defective states in solids could be a direct useful mean to improve the carrier's separation rate. This is because defects can be easily achieved just by controlling materials crystallinity. Most investigations have been performed to introduce surface defects that enhance the charge separation rate only referring to the organic photo-degradation. It is well established that the generation process of hydrogen and oxygen from water photolysis under light irradiation is totally different from the organic degradation process, because more requirement for semiconductors in water splitting than in organic photo-degradation. For instance, photocatalysts for water splitting have to possess a more negative conduction band bottom position than the redox potential of H+/H2 (0 eV vs. NHE), and the top position for valence has to be more positive than the redox potential of O2/H2O (1.23 eV vs. NHE).22 When defects are introduced as the electron traps onto materials surface, electron competitive migration could happen between electron trap level and conduction band, and therefore the competitive migration will influence the photocatalysts activity. Unfortunately, it still remains unclear how electron traps influence carrier's migration in water photolysis process. Therefore, it is highly important to study such an issue, most likely through a semiconductor-based hybridization via a systematic control over the defect amount.SnO2 is a prototype semiconductor with an appropriate energy band position (i.e., conduction band bottom at −0.11 eV and valence band top position at 3.59 eV)23 essential for photocatalytic water splitting, while it cannot response to visible light for its wide energy band gap. A recent study has demonstrated the potential of hybrid, g-C3N4/SnO2 as a hybrid photocatalyst for water splitting under visible light,24 which is based on the sole stoichiometric SnO2 without lattice defect. It is then very difficult to effectively tune the electron migration just via varying the corresponding stoichiometric content ratio in hybrid photocatalysts, and this kind of methods usually gives a single photocatalytic maximum, just referring to the electron migration between different photocatalysts' conduction bands. As a result, electron competitive migration mechanism caused by defects is poorly understood in hybrid photocatalysts. If defects are involved in the semiconductor-based hybrids, the relevant electron migration could be effectively tuned. It is highly possible to understand the photocatalytic mechanism in a systematic method which expects to promote the development of new photocatalysts and many other surface-sensitive technologies. We propose defective SnO2 as the target semiconductors to study, which was prepared via annealing of SnO2 quantum dots, since (i) SnO2 quantum dots can be easily fabricated, (ii) quantum dots usually have much of surface defect and low crystallinity just when prepared using SnCl4 hydrothermal method,25 and (iii) annealing of SnO2 quantum dots under given temperature could tune the defect concentration and vary the crystallinity. All these help one to find the appropriate methods of obtaining defective SnO2, with the amount of defects being regulated just by tailoring the mass ratio of SnO2.
Here, we initiated a systematic study on the electron competitive migration in defective tin dioxide by hybridizing with different amounts of polymer electron donor, g-C3N4. The amount of oxygen vacancy was tailored after subsequent solid calcinations. When tested as a photocatalyst, the hybrids showed dual maxima of visible-light catalytic activity in hydrogen generation from water under visible light irradiation, which totally differ from the only one catalytic activity maximum ever reported. The nature for this abnormal observation was comparatively studied by XPS, EPR and photoluminescence data in terms of an electron competitive migration mechanism.
2 Experimental
2.1 Materials
Melamine (C3H6N6), ethanol, SnCl4·5H2O and triethanolamine are chemically pure grade, all of the materials were purchased from Sinopharm Chemical Reagent Corp, P. R. China and used without further purification.2.2 Chemicals for materials preparation
Defective SnO2 nanocrystals were prepared via two steps. The first one involves the synthesis of SnO2 quantum dots with a poor crystallinity via a hydrothermal method. Namely, 0.5 mol SnCl4·5H2O was dissolved in 100 mL water and stirred for 10 min without adjusting pH value. This solution was poured into two 100 mL volume hydrothermal reactors with equal volume of 50 mL. Two reactors were then placed in an oven at 200 °C for 9 h. When the hydrothermal reactions finished, white slurry was obtained. After washing for several times using water, AgNO3 solution was used to detect the residual Cl−1 ions in supernatant till no evident white precipitate appeared. Then, the washed white precipitate was dried at 80 °C for 12 h, and milled into powder, which was named as SnO2-SH.
Further, we employed annealing process to prepare the lower defective SnO2 so that we can tailor the defect amount just simply changing the mass ratio of SnO2 essential for achieving the enhanced photocatalytic activity. For this purpose, SnO2-SH powders were annealed at 700 °C for 2 h in air at heating rate at 10 K min−1. With this process, the lower defective SnO2, named as SnO2-AD, was obtained. The amounts of oxygen vacancies were confirmed by XPS and EPR characterization.
2.3 Analytical and testing instruments
The particle morphologies of the samples were performed via transmission electron microscopy (TEM, JEM-2010 produced by JEOL). The phase structure and crystalline of samples were determined by X-ray diffraction patterns that were recorded on a Rigaku MinFlex II benchtop X-ray diffractometry (XRD) with Cu Kα irradiation (λ = 1.5418 Å). Thermogravimetric (TG) data of the samples were carried on Netzsch STA449F3 thermal analyzer. The chemical states of the samples were analyzed using X-ray photoelectron spectroscopy (XPS, a monochromatic Al Kα X-ray source operating at 150 W), and binding energies were referenced to the C1s peak at 284.6 eV that arises from adventitious carbon. Electron paramagnetic resonance (EPR) measurement for samples was performed via Bruker-BioSpin, E500 spectrometer to confirm the existence of oxygen vacancy. A frequency of ca. 9.866 GHz was used for a dual-purpose cavity operation. The magnetic field of 0.1 mT was modulated at 100 kHz. Fourier transform infrared (FT-IR) spectra of the samples were recorded on a Perkin-Elmer IR spectrometer using a KBr pellet technique. UV-vis absorbance spectra of the samples were measured by Varian Cary-500 spectrophotometer at ambient condition. Photoluminescence (PL) spectra in solid state and aqueous environment of the samples were carried out at room temperature by solid and aqueous Varian Cary Eclipse Fluorescence Spectrometers. During the aqueous environment PL tests, all samples with the same mass concentration must keep as their suspension state.2.4 Photocatalytic activity measurements
Photocatalytic hydrogen evolution was conducted in an online photocatalytic hydrogen generation system (Labsolar-IIIAG, Perfectlight Corp, Beijing, P. R. China) at a temperature of 5 °C. A 300 W Xe lamp equipped with a UV-cutoff filter (λ > 420 nm) was used as the light source of Sn–Hx hybrids, g-C3N4, SnO2-AD, and SnO2-SH/g-C3N4 hybrid in 40% SnO2 mass ratio while the light-cutoff filter (λ > 260 nm) was used alone in the hydrogen generation test of SnO2-AD to make sure its photocatalytic ability in hydrogen generation from water splitting. In details, in the photocatalytic process, 100 mg samples and 90 mL deionized water were added into the reaction reactor. 10 mL triethanolamine which acted as a sacrificial agent were added. The H2PtCl6 solution (1.0 wt% for Pt) was used for Pt deposition. The Pt would act as a co-catalyst to improve the photocatalytic performance of all samples. Prior to the reaction, the whole reaction system was deaerated to remove O2 and CO2. Gas evolution was analyzed by an on-line gas chromatograph (Fuli 9790 II) with TCD detector purchased from Fuli Analytical Instrument Corp, Zhejiang, P. R. China. Hydrogen evolution value is calculated based on 100 mg catalyst. Before the formal test, 30 minutes were spent for the light deposition of Pt from H2PtCl6, the deposition process could be described as follows:| H2PtCl6 → 2H+ + PtCl62− | (1) |
| PtCl62− + 4e− → Pt + 6Cl− | (2) |
Then the system was evacuated again to avoid the effect of the hydrogen ions of H2PtCl6, till it does not influence the hydrogen evolution from water splitting. The optical conversion capacity of g-C3N4 was defined by the photon–hydrogen conversion efficiency, as presented by the yield of hydrogen divided by g-C3N4 mass ratio in SnO2/g-C3N4 hybrids.
3 Results and discussion
3.1 Hybrids of defective SnO2 with g-C3N4: formation and characterization
 | ||
| Fig. 1 XRD patterns of sample SnO2-SH prepared directly by hydrothermal reaction, SnO2-AD obtained by annealing SnO2-SH at 700 °C for 2 h, g-C3N4 and selected hybrids Sn–Hx. | ||
After annealing quantum dots SnO2-SH at 700 °C for 2 h, the diffraction peaks became sharper and more intensive as for sample SnO2-AD in Fig. 1, which indicates an enhanced crystallinity and bigger grain size. The enhanced crystallinity would reduce the concentration of defects. It should be noted that calcinations at 700 °C did not alter the phase structure of nanocrystals, because the diffraction pattern of sample SnO2-AD after 700 °C calcinations also matched well with the standard diffraction data of rutile SnO2 (JCPDS, no. 41-1445). But its mean grain diameter grew up to 18.0 nm.
The as-prepared g-C3N4 shows two diffraction peaks at two theta of 13.0° and 27.7°, which correspond to (100) in-layer structural packing and (002) interlayer-stacking, respectively.5 The formation of hybrids between SnO2 nanoparticles and g-C3N4 was displayed in Fig. 1. It is clear that two sets of diffraction peaks for Sn–Hx hybrids are ascribed to SnO2 and g-C3N4, respectively. Because the main peaks for g-C3N4 and SnO2 are close so that double peaks in the 2θ range from 26.0° to 29.0° are overlapped for Sn–H5. Even so, careful data analyses indicate that the overlapped peaks could be distinguished to be associated with (110) diffraction of SnO2 and (002) peak of g-C3N4. With increasing the mass ratio of SnO2-AD, the diffraction intensity from SnO2-AD became increased (Fig. 1 and S2†). Furthermore, TG (Fig. S3†) measurements presented that Sn–H5, Sn–H30 and Sn–H60 samples have weight losses of about 95%, 70% and 40%, respectively, confirmed the mass ratios of defective SnO2 in the corresponding hybrids is compared with our started materials. The formation of Sn–Hx hybrids is also confirmed by FT-IR spectra (Fig. S4†). Different from only one wide and strong absorption for SnO2-AD appeared at 614 cm−1 in the frequency range of 400 to 2000 cm−1, hybrid samples exhibit three group peaks in the range of 400–800, 800–1000 and above 1000 cm−1. The broad weak peaks at low frequency are attributed to Sn–O–Sn vibration, while the other peaks are characteristic vibration of g-C3N4.
 | ||
| Fig. 2 TEM and HR-TEM images for samples: (A and B) Sn–H10, and (C and D) Sn–H40, profiles of lattice fringes for as-prepared SnO2-AD in (E) Sn–H10 hybrid and (F) Sn–H40 hybrid. | ||
As illustrated in Fig. 3a, two broad asymmetric photoelectron peaks centred at 486.3 eV and 494.7 eV were observed in Sn3d5/2 and Sn3d3/2 region, very closer to those of tin oxide.29 When carrying on the deconvolution of Sn3d, three principles should be followed: (i) the background was corrected according to the smart model; (ii) the peak area ratio of 3d3/2 to 3d5/2 was fixed at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 with a spin–orbital splitting distance of ca. 8.4 eV; and (iii) the FWHM of Sn3d5/2 component should be not more than 1.6 eV. Data analyses showed that signal Sn3d is consisted of three components: the strong one at 486.3 eV and 494.7 eV are attributed to Sn4+ ions, while that at 484.9 eV and 493.3 eV to Sn2+ ions that might be induced by oxygen vacancy (VO) via electron transfer from VO to Sn4+ atom.30 The third one Sn(X) at higher binding energy of 487.7 eV is still unknown, which may be caused by residual chloridion in SnO2-AD. Based on the peak areas as listed in Table 1, the relative ratio of [Sn2+] to [Sn4+] for Sn–H40 hybrid is about 6%.
:3 with a spin–orbital splitting distance of ca. 8.4 eV; and (iii) the FWHM of Sn3d5/2 component should be not more than 1.6 eV. Data analyses showed that signal Sn3d is consisted of three components: the strong one at 486.3 eV and 494.7 eV are attributed to Sn4+ ions, while that at 484.9 eV and 493.3 eV to Sn2+ ions that might be induced by oxygen vacancy (VO) via electron transfer from VO to Sn4+ atom.30 The third one Sn(X) at higher binding energy of 487.7 eV is still unknown, which may be caused by residual chloridion in SnO2-AD. Based on the peak areas as listed in Table 1, the relative ratio of [Sn2+] to [Sn4+] for Sn–H40 hybrid is about 6%.
 | ||
| Fig. 3 Core level spectra of (a) Sn3d and (b) O1s for Sn–H40. C1s and N1s recorded for sample g-C3N4 and Sn–H40 hybrid are compared in (c) and (d). | ||
| Peak components | Sn3d3/2 | Sn3d5/2 | O1s | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sn2+ | Sn4+ | Sn(X) | Sn2+ | Sn4+ | Sn(X) | H2Oads | ·OHads | O–Sn4+ | O–Sn2+ | |
| B.E. (eV) | 493.3 | 494.7 | 496.1 | 484.9 | 486.3 | 487.7 | 532.7 | 531.5 | 530.2 | 529.6 |
| FWHM (eV) | 1.44 | 1.41 | 1.58 | 1.58 | 1.44 | 1.58 | 1.50 | 1.57 | 1.43 | 1.25 |
| Peak area | 7519 | 168136 |
14840 |
11245 |
249251 |
22857 |
7263 | 21763 |
65765 |
1421 |
O1s core level was deconvoluted into four sub-peaks (Fig. 3b). One at the lowest binding energy of 529.6 eV should be corresponding to the oxygen species coordinated with Sn2+, while that at 530.2 eV to the oxygen species coordinated with Sn4+. The other two components with higher binding energies of 531.5 and 532.6 eV are associated with the surface species, such as tin(II) hydroxychloride and surface adsorbed water.31 The relative intensity ratio of the component at 529.6 eV to that at 530.2 eV is about 3%. C1s and N1s core levels of pure g-C3N4 and Sn–H40 were comparatively studied. In Fig. 3c, the signals at 287.2 and 288.0 eV for pure g-C3N4 are well attributed to carbon species of C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. Comparatively, both signals shifted towards 286.6 eV and 288.1 eV for Sn–H40. For N1s spectra in Fig. 3d, pure g-C3N4 exhibited three nitrogen species: the main signal at 398.2 eV is corresponding to the N atom with sp2 hybrid in N–CN, while that at 399.5 eV to N atoms in N–(C)3. The last one at higher binding energy of 400.6 eV contributes from the N species in N–H structure. Owing to the charging effects or positive charge localization in heterocycles, this peak was also observed at 404.3 eV, as reported elsewhere.32 For Sn–H40, N1s signals from N–CN and N–(C)3 shifted to 398.4 eV and 399.4 eV, respectively. The slight binding energy shifts of C1s and N1s indicated that a weak chemical interaction was formed between g-C3N4 and SnO2-AD.
N. Comparatively, both signals shifted towards 286.6 eV and 288.1 eV for Sn–H40. For N1s spectra in Fig. 3d, pure g-C3N4 exhibited three nitrogen species: the main signal at 398.2 eV is corresponding to the N atom with sp2 hybrid in N–CN, while that at 399.5 eV to N atoms in N–(C)3. The last one at higher binding energy of 400.6 eV contributes from the N species in N–H structure. Owing to the charging effects or positive charge localization in heterocycles, this peak was also observed at 404.3 eV, as reported elsewhere.32 For Sn–H40, N1s signals from N–CN and N–(C)3 shifted to 398.4 eV and 399.4 eV, respectively. The slight binding energy shifts of C1s and N1s indicated that a weak chemical interaction was formed between g-C3N4 and SnO2-AD.
 located at the surface of the target oxides.30 It should be noted that the electron traps (ETs) caused by oxygen vacancy in SnO2 has an important influence on the electron migrate property tailoring in hybrids. It is obvious that EPR signal intensity of SnO2-AD is stronger than that of SnO2 annealed at 900 °C, which indicates amount of oxygen vacancies defects decreased with increasing the annealing temperature in air.
located at the surface of the target oxides.30 It should be noted that the electron traps (ETs) caused by oxygen vacancy in SnO2 has an important influence on the electron migrate property tailoring in hybrids. It is obvious that EPR signal intensity of SnO2-AD is stronger than that of SnO2 annealed at 900 °C, which indicates amount of oxygen vacancies defects decreased with increasing the annealing temperature in air.
 | ||
| Fig. 4 EPR curves of the as-prepared sample SnO2-AD and the reference SnO2 annealing in 900 °C for 2 hours. | ||
 | ||
| Fig. 5 (A) Comparison of UV-vis absorbance spectra for sample SnO2-AD, g-C3N4 and selected Sn–Hx, and (B) calculation of energy band gap for samples g-C3N4 and SnO2-AD. | ||
3.2 Dual maxima of photocatalytic activity for the hybrids
So far, many experiments have proved that most of the hybrid photocatalysts, consisting of different semiconductors, give the higher photocatalytic activity comparing to their corresponding components. The basic enhancement mechanism is ascribed to the improvement of the photo-induced charges separation via electron migration among different semiconductors. Even so, majority of these hybrids usually exhibit only one photocatalytic activity maximum when tailoring the mass ratio of the component semiconductors. If surface defects are introduced as the charge trap centres onto one of component surfaces in hybrid photocatalysts, the competition for photo-generated electrons between conduction band and charge trap centre will be expected, which could also affect the photocatalytic activity in water photolysis. As a consequence, the photocatalytic activity may show a different tendency with two or more activity maximum. More importantly, it may bring us new understanding about electron transportation mechanism in hybrid photocatalysts. In the following, we initiated a study on the photochemical behaviour of hybrids that are constructed by defective SnO2 (SnO2-AD) and g-C3N4 at different SnO2 mass ratios, with a hope to get an efficient hydrogen generation from water splitting under visible light irradiation (λ > 420 nm).As shown in Fig. 6A and Table S1,† pure g-C3N4 deposited by 1 wt% Pt showed a hydrogen yield of 8.37 μmol when irradiated under visible light for 5 h. When g-C3N4 was hybridized with SnO2-AD, the hydrogen yield increased. Under the same test condition, Sn–H10 gave a first activity maximum with a hydrogen yield of 16.29 μmol, almost double of that for pure-C3N4. Further increasing the content of SnO2-AD led to the decrease in photocatalytic activity, as indicated in Fig. 6A. For Sn–H30, the hydrogen yield decreased to 7.74 μmol, a minimum value among all hybrids. Beyond 30% content of SnO2-AD, hydrogen yield of hybrids rose again and reached to 13.38 μmol for Sn–H40, the second activity maximum. That is, two photocatalytic activity maxima were presented for SnO2-AD/g-C3N4 hybrids by tailoring the mass ratio of SnO2-AD. It should be emphasized that, the activity of as-prepared SnO2 in water photolysis is really poor to yield hydrogen of 3 μmol and 0 μmol (Fig. S8†) after irradiation for 5 h under the ultraviolet (λ > 260 nm) and visible (λ > 420 nm) light even though it had appropriate energy band position as well as narrower gap (3.5 eV). To confirm the negative role of oxygen vacancies in SnO2/g-C3N4 hybrids, the hybrid in 40% mass ratio of SnO2-SH was also tested in the same condition, the results indicated that the photocatalytic activity became weakened when compared with that of Sn–H40 (Fig. S9†). Such weakening in photocatalytic activity of hybrid in water splitting confirms that there should be a competitive migration between conduction band and oxygen vacancy.
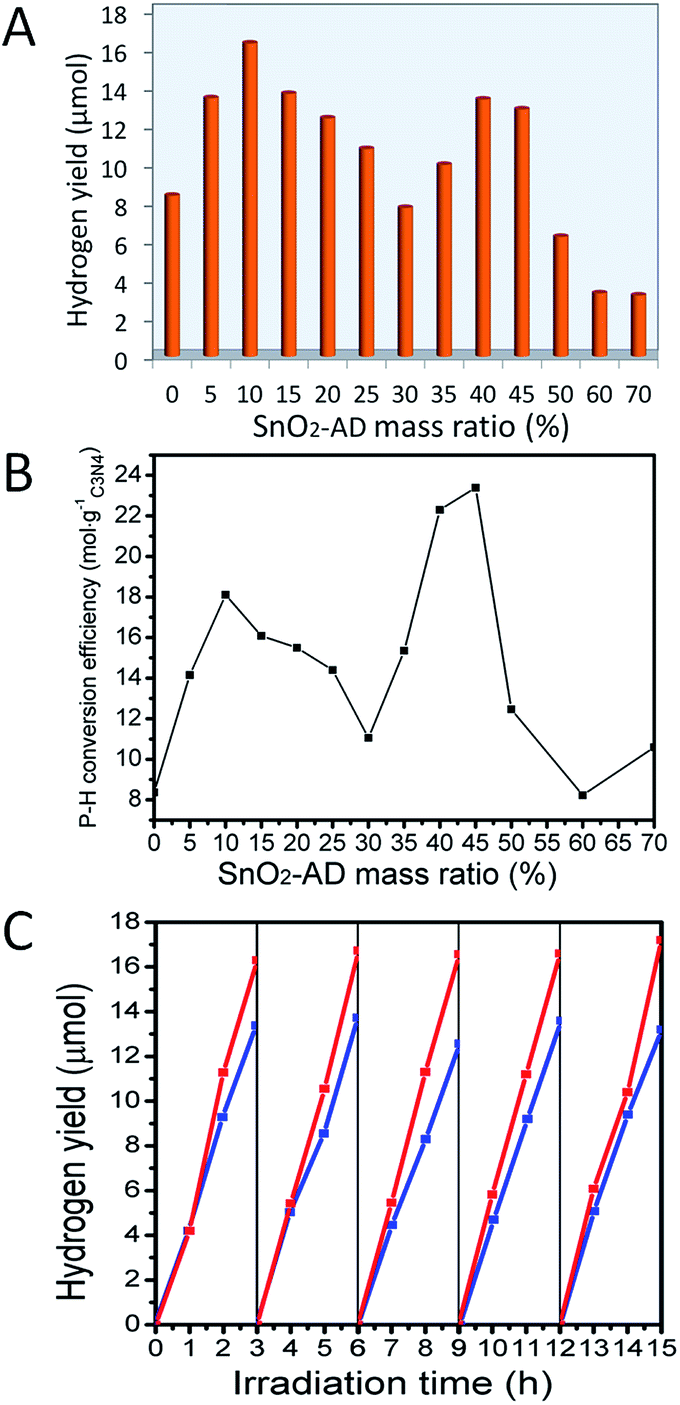 | ||
| Fig. 6 (A) Variation of hydrogen yields from water splitting under visible light (420 nm) irradiation for 5 h with varying SnO2-AD content in Sn–Hx hybrids; (B) photon–hydrogen (P–H) conversion efficiency of these hybrids; and (C) comparison of the photocatalytic stability for sample Sn–H10 (red lines) and Sn–H40 (blue lines). | ||
As displayed in Fig. 6B, the photon–hydrogen (P–H) conversion efficiency of hybrids also showed two peaks, i.e., Sn–H10 gave the first P–H conversion efficiency peak of 14.15 μmol gC3N4−1, and the second one of 23.39 μmol gC3N4−1 belongs to Sn–H45, also the highest conversion efficiency value among all hybrids. Sn–H30 and Sn–H60 exhibited P–H conversion efficiency minima. The high SnO2-AD content dependence of photocatalytic activity for SnO2-AD/g-C3N4 hybrids, especially for dual maxima observed in hydrogen yield (Fig. 6A) and P–H conversion efficiency (Fig. 6B), should be associated with the trapping of excited electron by defects of SnO2-AD component.
In addition to the hydrogen yield and P–H conversion efficiency, photochemical stability is also a key factor in evaluating photocatalyst. As illustrated in Fig. 6C, photocatalytic activity of H2 yield for Sn–H10 and Sn–H40 hybrids can be maintained at the same level after 5 times cycle tests, demonstrating that the obtained SnO2-AD/g-C3N4 hybrids are stable in the photocatalytic process and could be potentially used in practical clean energy fields.
3.3 Origin of dual catalytic activity maxima
Introducing electron traps in SnO2 component results in dual activity maxima for SnO2–Hx hybrids. Therefore, there should be a different photo-generated carrier's migration mechanism from other routine hybrids with single maximum. As well known, water photolysis is a reaction that involves the contributions from photo-generated electrons/holes. The photo-generated charges have two ways of retuning back to the ground state: one is via the radiative recombination caused by electron–hole pair recombination. This process will release photon. Another one is non-radiative recombination via complex electron–phonon interaction, which would release heat. Photoluminescence (PL) test is often employed to evaluate the radiative recombination rate of photo-generated electrons/holes. Usually, the higher PL intensity indicates a higher radiative recombination rate of photo-generated charges under the same excitation wavelength, which means photo-generated charges cannot be separated completely. In the present investigation, PL spectra of hybrids were recorded and will be discussed as follows.Fig. 7A and S10A† displayed the PL spectra of samples measured for the solid state powder directly. All samples exhibit a wide band emission peaking at about 460 nm. As a whole, PL integral intensity decreased as SnO2-AD mass ratio increases (Fig. 7C), which means that SnO2-AD could efficiently improve the photo-generated charges separation rate, and PL delay measurement of g-C3N4 and SH-5 also approved that SnO2-AD can decrease the intrinsic charges recombination rate of g-C3N4 (see Fig. S12†). Pure g-C3N4 had the highest PL intensity among all samples, which is associated with its highest radiative recombination rate of photo-generated carries that govern its low P–H conversion efficiency in Fig. 6B. Even though sample Sn–H60 gave a weaker PL emission as indicated by its lower PL integral intensity (Fig. 7C), it did not display a high P–H conversion efficiency. This observation infers that photo-generated electrons in sample Sn–H60 do not take effectively part in the photochemical reaction. That is to say, photo-generated electrons could be trapped by oxygen vacancy in SnO2-AD. Moreover, these trapped electrons might deliver energy to lattice vibration via phonon-electron interactions rather than the recombination with holes via radiative emission. It should be mentioned that PL test for solid state powders can only partly reveal some information about carrier separation rate at the absence of solution environment.
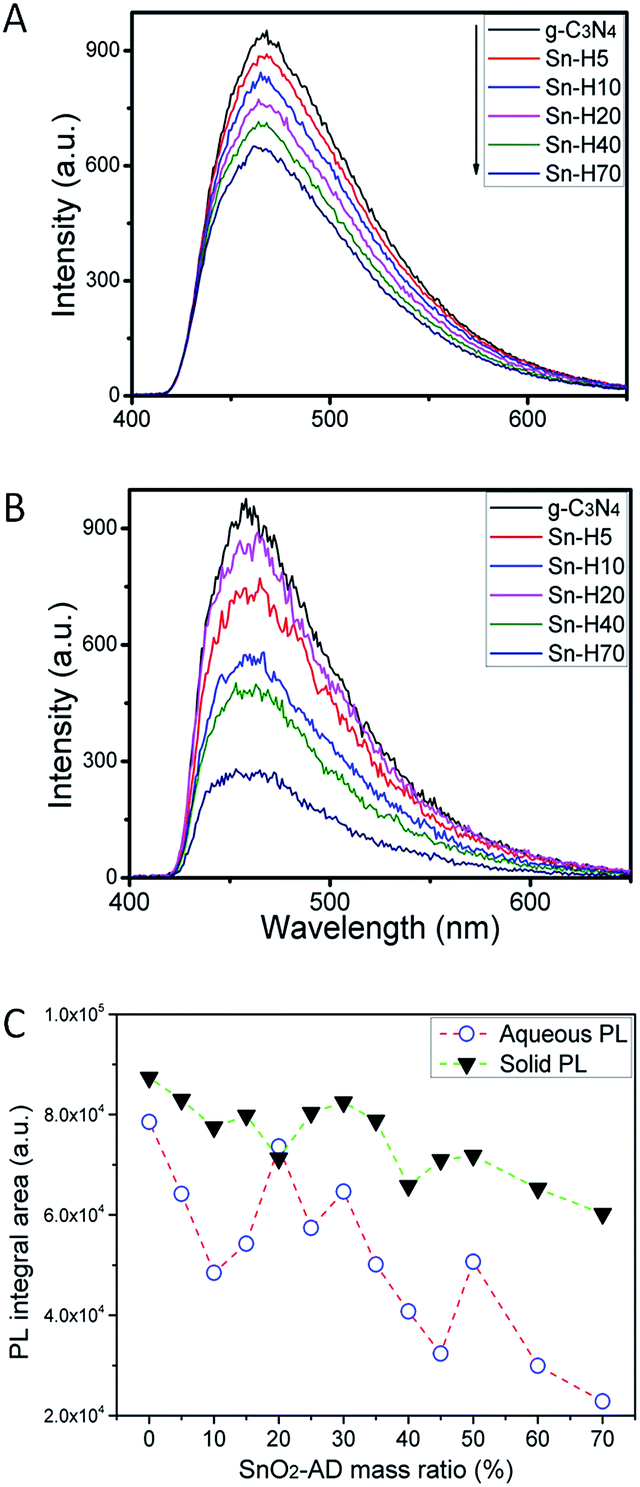 | ||
| Fig. 7 PL intensity spectra in (A) solid state and (B) aqueous state of g-C3N4, selected Sn–Hx (C) PL integral areas of Sn–Hx under 390 nm excitation wavelength. | ||
In water splitting experiment, photocatalyst participates in two photo-energy converter processes: one is the light-to-chemical photoenergy converters that yield hydrogen, and another one is light-to-light photoenergy converters that produce PL emission. Capturing PL signal in aqueous solution has been reported by Yeh et al.33 for understanding water splitting. In fact, water photolysis reaction occurs in aqueous environment. This aqueous state would provide a quasi-in situ environment owing to the co-existence of ·OH and H+ in solution. Even though the absence of co-catalysts and react agents in PL test might reduce the reaction rate of splitting water into hydrogen and oxygen, PL test in aqueous state, much different from the solid state emission, is still a effective method to give more detailed information about charge action among water photolysis reactions, especially for defective photocatalysts.
PL spectra recorded for Sn–Hx hybrids in suspension aqueous environment in the absence of sacrificial agent and co-catalyst are shown in Fig. 7B and S10B.† During the test, the mass concentration of sample was kept the same so as to evaluate PL intensity of hybrids. Three different features could be easily found for solution emission from those for solid state powders. Firstly, the intensity decreases rapidly with increasing SnO2-AD content when proceeding PL test in aqueous environment comparing to that in solid state when maintaining the test parameters, which means that more free electrons were separated by SnO2 in aqueous state than in solid state. Secondly, PL peak position exhibited apparent blue shifts from 464 nm in solid state emission to 458 nm in aqueous emission. One may thus expect that some other kinds of emissions in hybrids might contribute to the whole PL intensity except for those in g-C3N4, such as the interface recombination caused by oxygen defects.34 Thirdly, pure g-C3N4 and sample Sn–H70 gave the strongest and the weakest emission. Moreover, sample Sn–H30 exhibited much more intensive emission when comparing to the neighbouring composition (Fig. 7C). This observation is in good agreement with their P–H maxima. Even though the hybrid Sn–H20 has shown a higher aqueous PL intensity, excellent P–H conversion efficiency is still indicated, superior to those for Sn–H25 and Sn–H30. Therefore, there exist a higher radiative recombination rate of photo-generated carriers on the interface and lower electron–phonon interaction rate in oxygen vacancy. PL test in solid state and suspension environment displayed a paradox between PL intensity and P–H conversion efficiency. What should be noted, after deposited 1 wt% Pt for all of the samples, the PL spectra integral areas trend in triethanolamine solution environment is same to the PL properties in pure water (Fig. S11†). Which approved the deposition of Pt didn't change the PL properties trend in a very short period of time.
Combining with the PL properties of samples in solid and in aqueous environments, interface recombination and electron–phonon interaction caused by the electron trapping in oxygen vacancy would be expected to play a key role in photo-generated carrier separation rate. Therefore, the existence of oxygen vacancy in SnO2 nanocrystals should be the dominated factor for the dual maxima in hydrogen yield.
Here, we proposed an oxygen vacancy induced electron competitive migration mechanism in hybrids. It is well documented that photo-generated carrier migration between different semiconductors is determined by the energy band position of component in hybrid, which could be important in affecting photocatalytic activity. As illustrated in Scheme 1, electrons in valence band (VB) of SnO2 cannot be excited to the conduction band (CB) under visible-light irradiation due to a wide band gap (3.5 eV). All electrons that participated in the water photolysis reaction should come from g-C3N4. Under the irradiation of visible light, electrons are excited from the VB of g-C3N4 to the CB. These excited electrons would be divided into three parts: (i) the first part is those that participate directly in the reduction of H+ on g-C3N4 surface, (ii) the second part is those that move to the CB of SnO2-AD as free electron or trapping by defect level of oxygen vacancy in SnO2-AD as localized electron, and (iii) the third one is those that recombine with photo-generated holes via radiative or non-radiative transition. On the one hand, the lower valence band position of SnO2-AD prevents the transporting of holes from valence band of g-C3N4 to that of SnO2-AD. More importantly, less of photo-generated holes appear at VB of SnO2-AD because the transition of hole from VB of g-C3N4 to that of SnO2-AD is electrical-dipole-forbidden. Therefore, recombination possibility of electrons in the CB of SnO2-AD with holes would be greatly reduced. These photo-generated electrons move to the CB of SnO2-AD, and would also take part in the water splitting and enhance the photocatalytic activities.
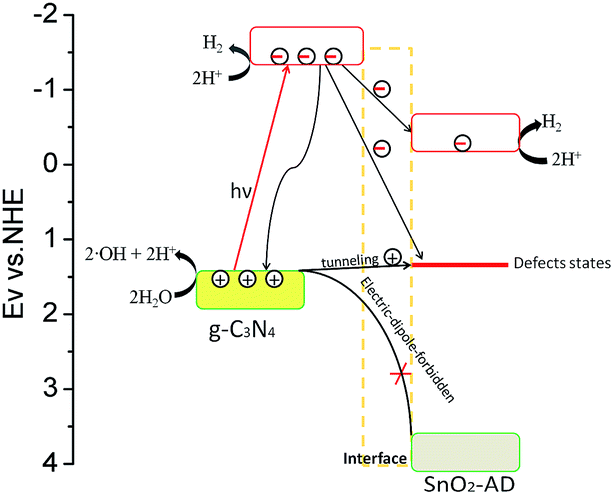 | ||
| Scheme 1 Relative band position of SnO2-AD/g-C3N4 hybrid photocatalyst, and the migration of photo-generated carriers in the hybrid. The trap level affected the carriers' migration between defective direct-gap semiconductor SnO2-AD and indirect-gap semiconductor g-C3N4. | ||
The electrons that are trapped by defect level (about 2.1 eV above value band top as reported by ref. 35) of oxygen vacancy could be consumed by two processes. In the first process, trapped electrons deliver their energy to the lattice vibration of SnO2-AD due to electron–phonon interaction, and in another one, recombining with holes from g-C3N4 because oxygen vacancy could become the recombination centre of photo-generated carrier.36 The coulomb attraction between trapped electrons and holes help them overcome the interface potential bias between g-C3N4 and SnO2-AD. Therefore, some of holes in g-C3N4 would be attracted to the adjacent defect position and recombine with trapped electrons on SnO2-AD/g-C3N4 interface. With increasing SnO2-AD mass ratio, more photo-generated electrons would be trapped in oxygen vacancy. In this regard, there might cause a competitive electron migration to SnO2-AD CB and defective level. When the content of SnO2-AD is less than 10% in hybrids, the electron migration just happen among different CB and interior of g-C3N4. Less of electrons would be trapped by defect level of oxygen vacancy for the little amount of SnO2-AD. Therefore, the variation of PL intensity detected for suspension environment is the same as the P–H conversion efficiency, and reaches the first photocatalytic maximum at Sn–H10. As more SnO2-AD component was introduced in hybrids, the amount of trapped electron increases obviously. The coulomb attraction between trapped electrons and holes become stronger at the same time, which resulted in more free holes tunnelling g-C3N4/SnO2-AD interface and recombining with trapped electrons via radiative recombination. Alternatively, the free photo-generated carriers in hybrids decease due to the reducing of g-C3N4 mass ratio. When SnO2-AD mass ratio reaches to 20%, the coulomb attraction would reach to a maximum, giving a higher interface radiative recombination rate. At this composition, the electron–phonon interaction is still weak, resulting in a higher aqueous PL intensity for Sn–H20 when comparing to Sn–H25 and Sn–H30. As SnO2-AD mass ratio increases beyond 30%, the interface recombination start to decrease evidently, and electron–phonon interaction becomes the predominant energy delivering process for the trapped electron. Owing to the limited recombination rate, the second maximum in photocatalytic activity is still observed for Sn–H45.
4 Conclusions
Defective SnO2 nanoparticles were fabricated via two steps including hydrothermal reaction and subsequent annealing. By tailoring the mass ratio of defective SnO2 in SnO2/g-C3N4 hybrids, we investigated the influence of oxygen vacancy on the photo-induced carries competitive migration and photocatalytic activity of hybrids. HR-TEM, XPS and EPR test approved that oxygen vacancies exist on the surface of SnO2, which acted as the electron trap centre and resulted in competitive electron migration of photo-generated carriers in defective SnO2. Comparing to other routine hybrids with single photocatalytic activity maximum, double peak value were observed in water splitting test for SnO2/g-C3N4 hybrids, and the hybrids with SnO2 relative mass ratio of 10 wt% and 40 wt% showed double peak hydrogen generation rate of 54.3 μmol h−1 g−1 and 44.3 μmol h−1 g−1, both are far beyond of pure g-C3N4 (27.9 μmol h−1 g−1) under visible light irradiation. Photon–hydrogen conversion efficiency also showed two maxima by tailoring the defective SnO2 weight ratio. This work develops a method that regulates oxygen vacancy amount by tailoring defective SnO2 content essential for comprehending the impacts of oxygen vacancies on photocatalytic activity. Moreover, the relevant experimental results demonstrate that the electron competitive migration is caused by oxygen vacancy in water photolysis. Therefore, the findings reported in this work could be highly helpful to deeply understand oxygen vacancy and its impacts in hybrids, and hopefully to give a new idea in designing novel photocatalysts for water splitting.Acknowledgements
This work was financially supported by NSFC (21025104, 21271171, 21401190 and 91022018).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- Y. Tang, H. Zhou, K. Zhang, J. Ding, T. Fan and D. Zhang, Chem. Eng. J., 2015, 262, 260–267 CrossRef CAS.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y.-H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- W. Liu, M. Wang, C. Xu, S. Chen and X. Fu, J. Mol. Catal. A: Chem., 2013, 368, 9–15 CrossRef.
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168, 1–8 Search PubMed.
- J. Zhang, Y. Wang, J. Jin, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 10317–10324 CAS.
- Z. L. Fang, H. F. Rong, Z. L. Ya and P. Qi, J. Mater. Sci., 2015, 50, 3057–3064 CAS.
- Y. Xu and W. D. Zhang, Eur. J. Inorg. Chem., 2015, 1744–1751 CrossRef CAS.
- J. Shen, H. Yang, Q. Shen, Y. Feng and Q. Cai, CrystEngComm, 2014, 16, 1868–1872 RSC.
- Y. Zang, L. Li, Y. Xu, Y. Zuo and G. Li, J. Mater. Chem. A, 2014, 2, 15774–15780 CAS.
- H. Chen, Y. Xie, X. Sun, M. Lv, F. Wu, L. Zhang, L. Li and X. Xu, Dalton Trans., 2015, 44, 13030–13039 RSC.
- J. Lei, Y. Chen, L. Wang, Y. Liu and J. Zhang, J. Mater. Sci., 2015, 50, 3467–3476 CAS.
- W. Li, C. Li, B. Chen, X. Jiao and D. Chen, RSC Adv., 2015, 5, 34281–34291 RSC.
- L. Huang, H. Xu, Y. Li, H. Li, X. Cheng, J. Xia, Y. Xu and G. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC.
- S. Chen, Y. Hu, S. Meng and X. Fu, Appl. Catal., B, 2014, 150, 564–573 CrossRef.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS.
- Y. Su, J. Lang, N. Cao, T. Wang, B. Zhu and X. Wang, J. Nanopart. Res., 2015, 17, 63 CrossRef.
- J. Zhang, J. H. Bang, C. Tang and P. V. Kamat, ACS Nano, 2010, 4, 387–395 CrossRef CAS PubMed.
- A. Suryawanshi, P. Dhanasekaran, D. Mhamane, S. Kelkar, S. Patil, N. Gupta and S. Ogale, Int. J. Hydrogen Energy, 2012, 37, 9584–9589 CrossRef CAS.
- R. Yin, Q. Luo, D. Wang, H. Sun, Y. Li, X. Li and J. An, J. Mater. Sci., 2014, 49, 6067–6073 CrossRef CAS.
- Y. Zang, L. Li, X. Li, R. Lin and G. Li, Chem. Eng. J., 2014, 246, 277–286 CrossRef CAS.
- Y. Zhang, L. Li, J. Zheng, Q. Li, Y. Zuo, E. Yang and G. Li, J. Phys. Chem. C, 2015, 119, 19505–19512 CAS.
- A. Das, V. Bonu, A. K. Prasad, D. Panda, S. Dhara and A. K. Tyagi, J. Mater. Chem. C, 2014, 2, 164–171 RSC.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- G. S. Li, J. Boerio-Goates, B. F. Woodfield and L. P. Li, Appl. Phys. Lett., 2004, 85, 2059–2061 CrossRef CAS.
- A. W. C. Lin, N. R. Armstrong and T. Kuwana, Anal. Chem., 1977, 49, 1228–1235 CrossRef CAS.
- C. Canevali, N. Chiodini, P. DiNola, F. Morazzoni, R. Scotti and C. L. Bianchi, J. Mater. Chem., 1997, 7, 997–1002 RSC.
- C. Fettkenhauer, G. Clavel, K. Kailasam, M. Antoniettia and D. Dontsova, Green Chem., 2015, 17, 3350–3361 RSC.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. S. Teng, Adv. Mater., 2014, 26, 3297–3303 CrossRef CAS PubMed.
- T. V. Torchynska, A. D. Cano, M. M. Rodriguez and L. Y. Khomenkova, Phys. B, 2003, 340, 1113–1118 CrossRef.
- A. Kar, M. A. Stroscio, M. Dutta, J. Kumari and M. Meyyappan, Appl. Phys. Lett., 2009, 94, 101905 CrossRef.
- A. Kar, S. Kundu and A. Patra, J. Phys. Chem. C, 2011, 115, 118–124 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23361e |
| This journal is © The Royal Society of Chemistry 2016 |