DOI:
10.1039/C5RA23189B
(Paper)
RSC Adv., 2015,
5, 103082-103090
Effect of carbon content and electronic strong correlation on the mechanical and thermodynamic properties of ytterbium carbides
Received
4th November 2015
, Accepted 24th November 2015
First published on 26th November 2015
Abstract
The mechanical and thermodynamic properties of four ytterbium carbides with increasing carbon content have been examined using ab initio calculations based on density functional theory. In order to describe the strong on-site Coulomb repulsion among localized 4f electrons, generalized gradient approximation plus a Hubbard parameter (GGA + U) formalisms have been adopted for the exchange correlation term. The elastic constants of YbC, Yb2C3, YbC2, and YbC6 are related to Hubbard U. The bulk modulus B, shear modulus G, and Young's modulus E are evaluated through Voigt–Reuss–Hill approximation. Among the four ytterbium carbides, YbC6 yields the largest B, G, and E, and YbC2 exhibits relatively soft and ductile characteristics. Mechanical anisotropy was estimated using several anisotropic indexes and factors. The anisotropic property of E of four ytterbium carbides is more evident than that of B. A phonon calculation reveals the thermodynamic stability of YbC2 and YbC6, which is consistent with experimental observations.
1. Introduction
Metal carbides have been extensively investigated from both experimental and theoretical points of view because of their unique magnetic and electrical properties for electronic applications.1–4 Rare-earth carbides have been commonly used as model systems to probe structure–property relationships because of their structural diversity. C atoms play a principal role in the structural diversity of rare-earth carbides. The C-to-metal atomic ratio in rare-earth carbides exhibits a broad range, for instance, the C-to-metal atomic ratios of CeC and EuC6 are 1 and 6, respectively.5–7 The C atoms in rare-earth carbides also exist in various forms, such as three-dimensional networks, two-dimensional layered structures, and C–C dumbbell units. Two-dimensional C layers are implicated in superconducting graphite intercalation compounds (GICs). The combination of the graphene and intercalated sheets is accounted for superconductivity because neither graphite nor pure metal exhibits a high Tc.8 It is reported that the C–C dumbbell distance is correlated with Tc of rare-earth carbides superconductors.9 The C–C distance in the C2 dumbbell at approximately 1.30 Å shows the highest Tc among rare-earth carbides. The C2 dumbbell is also found in RExTyC2 carbides, where RE denotes a rare-earth metal and T denotes a transition metal. C2 and T form a quasi-one-dimensional linear TC4 unit embedded in matrix RE cations. It is found that the medium-frequency C2 vibrations drive the high Tc in Sc3CoC4.10 Furthermore, C2 dumbbell dimer unit could transfer to two-dimensional graphite sheets under pressure, such as in Ca–C systems.11–13
The past decade has been seen intense efforts directed at studies of the structures and properties of ytterbium carbides. The ytterbium–carbon systems are significantly different from those of lightweight rare-earth carbide systems, which have well defined dicarbides, Pu2C3-type sequicarbides, and NaCl-type monocarbides.14,15 However, NaCl-type monocarbides with a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 has yet to be determined in Yb–C systems. Yb3C is characterized by a C-deficient NaCl-type structure.14 Although Yb2C3 with a body-centered cubic Pu2C3-type structure can be prepared at high pressures, the thermodynamic stability of this compound under ambient conditions remains elusive.16 Similar to lightweight rare-earth carbides, YbC2 assumes a tetragonal CaC2-type structure.17 Surprisingly, the lattice parameters of YbC2 are different from other rare-earth dicarbides. The lattice parameters of rare-earth carbides decrease regularly with increasing of atomic number of metal atoms, by contrast, the lattice parameters of YbC2 lie between those of HoC2 and ErC2.14 The interest of ytterbium carbides is in part fueled by the discovery of superconductivity in YbC6 with Tc of 6.5 K.18 Two mechanisms have been proposed to understand the superconductivity of GICs, including YbC6. Csányi et al. proposed an unconventional exciton or plasmon-mediated pairing mechanism.19 Later, a conventional BCS phonon-mediated mechanism has also been suggested; in this mechanism, interlayer bands display a sufficiently strong coupling with both in-plane intercalant and out-of-plane graphite phonon modes to allow for a relatively high Tc.20,21
:1 has yet to be determined in Yb–C systems. Yb3C is characterized by a C-deficient NaCl-type structure.14 Although Yb2C3 with a body-centered cubic Pu2C3-type structure can be prepared at high pressures, the thermodynamic stability of this compound under ambient conditions remains elusive.16 Similar to lightweight rare-earth carbides, YbC2 assumes a tetragonal CaC2-type structure.17 Surprisingly, the lattice parameters of YbC2 are different from other rare-earth dicarbides. The lattice parameters of rare-earth carbides decrease regularly with increasing of atomic number of metal atoms, by contrast, the lattice parameters of YbC2 lie between those of HoC2 and ErC2.14 The interest of ytterbium carbides is in part fueled by the discovery of superconductivity in YbC6 with Tc of 6.5 K.18 Two mechanisms have been proposed to understand the superconductivity of GICs, including YbC6. Csányi et al. proposed an unconventional exciton or plasmon-mediated pairing mechanism.19 Later, a conventional BCS phonon-mediated mechanism has also been suggested; in this mechanism, interlayer bands display a sufficiently strong coupling with both in-plane intercalant and out-of-plane graphite phonon modes to allow for a relatively high Tc.20,21
4f electrons play a pivotal role in understanding the electronic and thermodynamic properties of rare-earth compounds; however, conventional density function theory (DFT), which uses conventional exchange-correlation potential, such as local density functional approximation (LDA) or generalized gradient approximation (GGA), has failed to describe the ground state properties of systems containing 4f electrons because of their strong on-site Coulomb repulsion. An effective modification of pure DFT which calls LDA/GGA + U (U is the Hubbard energy) scheme22–24 can be used to obtain considerable improvement with respect to the LDA or GGA results. In fact, DFT + U calculations have been performed to explore the electronic properties of ytterbium compounds. Including localized 4f orbitals in the LDA + U method results in the downward rigid shift of the energy of the filled 4f bands in YbC6.25 The more recently studies of novel correlated topological insulator YbB6 reveals that the 4f band moves to a higher energy and does not cross the 5d band when turning on the Hubbard parameter U; this findings is consistent with experimental data.26
Although numerous studies on the structural and electronic properties of individual ytterbium carbide have been performed, theoretical studies on ytterbium carbides, especially mechanical and thermodynamic properties with respect to carbon contents have been rarely conducted. In addition, the effects of electronic strong correlation on ytterbium carbides have been sparsely visited. In this work, four representative ytterbium carbides with increasing carbon contents (YbC, Yb2C3, YbC2, and YbC6) were selected to systematically examine their mechanical and thermodynamic properties through ab initio GGA + U calculations.
2. Computational methods
The ab initio calculations were performed within the framework of DFT as implemented in the Vienna Ab-initio Simulation Package (VASP).27 The all-electron projector augmented wave (PAW)28 pseudopotential for Yb and C from the VASP pseudopotential library were used. The electron wave function was expanded in plane waves up to a cutoff energy of 550 eV. The k-point meshes29 in the full edge of the Brillouin zone (BZ) are sampled of 2π × 0.02 Å−1. Iteration relaxation of atomic positions and cell volume was stopped when the forces generally acting on the atoms were found to be smaller than 0.01 eV Å−1. With this criterion, the change in total energy between successive steps was less than 0.01 meV per cell. The exchange and correlation energy was assessed by GGA in the scheme of Perdew–Burke–Ernzerhof (PBE).30 The strong on-site Coulomb repulsion among the localized Yb 4f electrons was described by the DFT + U method22,24 developed by Dudarev et al.23 In this scheme, the total GGA energy functional is defined as:
where ρσ is the density matrix of f states with spin σ, U and J are the spherically averaged screened Coulomb energy and exchange energy, respectively. Since only the difference between U and J is significant, thus we will henceforth label them as one single parameter U for simplicity, In the subsequent calculations, the parameter U varies from 1 to 5, and U = 0 means turning off the Hubbard U term.
Phonon calculations were performed by supercell approach and finite displacement method.31 From finite displacements, the Hellmann–Feynman atomic forces computed at the optimized supercell by VASP code were transferred to the PHONOPY code32,33 to construct the dynamical matrix. Diagonalization of the dynamical matrix gives phonon frequencies and their corresponding density of states. In the interpolation of the constants for calculating the phonon dispersion, 3 × 3 × 3, 2 × 2 × 2, 3 × 3 × 3, and 2 × 2 × 1 supercells with k-point mesh of 2π × 0.02 Å−1 were used for YbC, Yb2C3, YbC2 and YbC6 respectively. Thermodynamic properties can be determined by phonon calculations using the quasiharmonic approximation (QHA).33 The phonon contribution to the Helmholtz free energy F is given by:
where
ω(
q,
v) is the phonon frequency at wave vector
q and band
v, and
T is the temperature.
kB and
ℏ are the Boltzmann constants and the reduced Planck constants, respectively. The heat
CV and
S are given by
and
respectively.
3. Results and discussion
3.1 Equilibrium structural properties
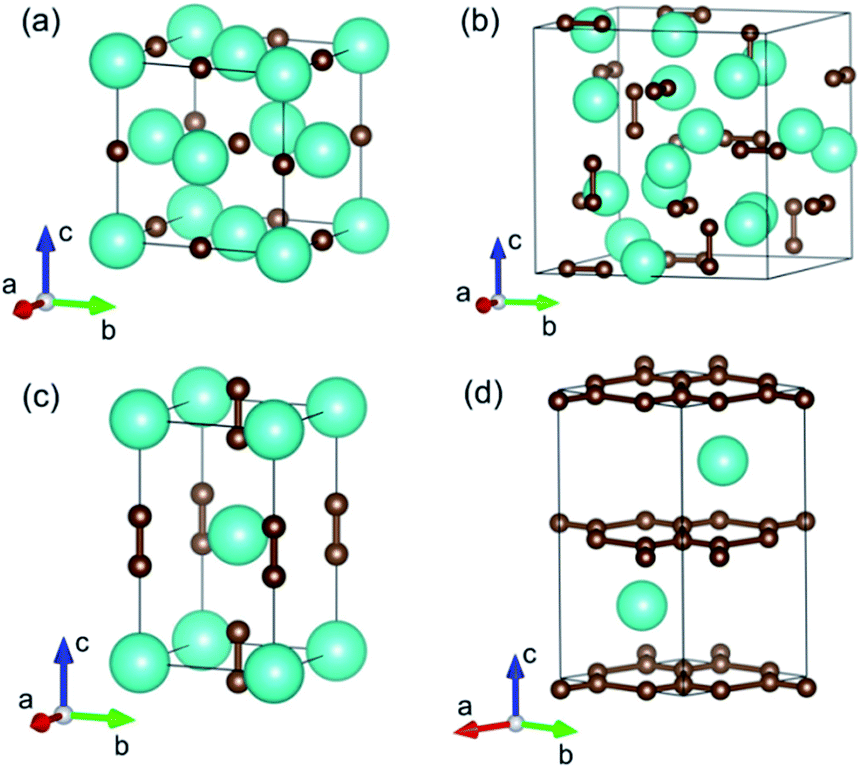
YbC with a stoichiometric ratio of 1:1 adopts an ordered cubic NaCl-type structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), in which Yb atoms assume Na positions and C atoms occupy Cl positions, as shown in Fig. 1(a). The Yb atoms form a close-packed structure, whereas the C atoms enter octahedral interstices. The lattice constant a of YbC with various Hubbard U values are presented in Fig. 2(a). The lattice constants display a ladder shape as Hubbard U values increase. When Hubbard U is turned off, the calculated lattice constant a reaches 5.024 Å, which is the smallest value. At Hubbard U = 5 eV, the lattice constant a is 5.062 Å. Ytterbium sequicarbides, Yb2C3, assumes a cubic Pu2C3-type crystal structure (space group I
m), in which Yb atoms assume Na positions and C atoms occupy Cl positions, as shown in Fig. 1(a). The Yb atoms form a close-packed structure, whereas the C atoms enter octahedral interstices. The lattice constant a of YbC with various Hubbard U values are presented in Fig. 2(a). The lattice constants display a ladder shape as Hubbard U values increase. When Hubbard U is turned off, the calculated lattice constant a reaches 5.024 Å, which is the smallest value. At Hubbard U = 5 eV, the lattice constant a is 5.062 Å. Ytterbium sequicarbides, Yb2C3, assumes a cubic Pu2C3-type crystal structure (space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d) with eight formula units (fu) in a conventional cell. The Yb atoms are aligned along the [111] direction, and the C atoms form dumbbell-shaped dimers occupying the voids in the bisphenoids of the Yb substructure. The calculated lattice constant a as a relationship of Hubbard U is shown in Fig. 2(b). The calculated lattice constant a of 8.290 Å at Hubbard U = 0 eV is 2.7% larger than the experimental value of 8.073 Å.16 Furthermore, the calculated C–C distances slightly decrease as Hubbard U value increases, from 1.314 Å at Hubbard U = 0 eV to 1.303 Å at Hubbard U = 5 eV. However, the difference between the calculated C–C distances with various Hubbard U values within 0.2% and the C–C dimer distances of Yb2C3 are close to the experimental value of 1.313 Å.16
3d) with eight formula units (fu) in a conventional cell. The Yb atoms are aligned along the [111] direction, and the C atoms form dumbbell-shaped dimers occupying the voids in the bisphenoids of the Yb substructure. The calculated lattice constant a as a relationship of Hubbard U is shown in Fig. 2(b). The calculated lattice constant a of 8.290 Å at Hubbard U = 0 eV is 2.7% larger than the experimental value of 8.073 Å.16 Furthermore, the calculated C–C distances slightly decrease as Hubbard U value increases, from 1.314 Å at Hubbard U = 0 eV to 1.303 Å at Hubbard U = 5 eV. However, the difference between the calculated C–C distances with various Hubbard U values within 0.2% and the C–C dimer distances of Yb2C3 are close to the experimental value of 1.313 Å.16
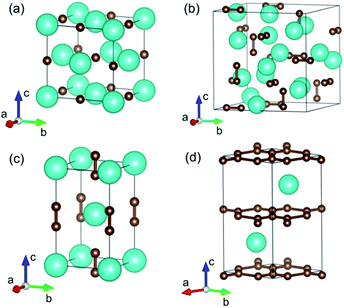 |
| | Fig. 1 Crystal structures of (a) YbC, (b) Yb2C3, (c) YbC2, and (d) YbC6. The large and small spheres represent Yb and C atoms, respectively. | |
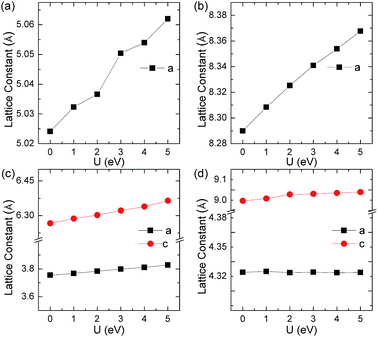 |
| | Fig. 2 Lattice constants as a function of Hubbard U for (a) YbC, (b) Yb2C3, (c)YbC2, and (d) YbC6. | |
YbC2 crystallizes in a body-centered tetragonal CaC2-type structure (space group I4/mmm), in which the C atoms exist in the form of a C–C dimer similar to that in Yb2C3. The C–C dimers oriented parallel to the c axis are six-fold coordinated by Yb atoms forming an elongated octahedron. As the Hubbard U increases from 0 eV to 5 eV, the lattice constants a and c increase by 1.9% and 1.6%, respectively. The calculated C–C dimer distances in YbC2 with various Hubbard U remain almost unchanged and reach the experimental value of 1.293 Å.17 Moreover, it is noteworthy that the C–C dimer distance in other Pu2C3- and CaC2-type rare-earth carbides is approximately 1.3 Å, such as 1.298 Å in Y2C3,34 1.2942 Å in La2C3,35 1.2888 Å in YC2,36 and 1.303 Å in LaC2.37 This result indicates that the C–C dimer distance shows a weak dependence on the choice of metal atoms in these crystals.
Thus, it is not unexpected that the C–C dimer distances in Yb2C3 and YbC2 change slightly with various Hubbard U because the Hubbard U correction is added on the 4f electrons of the Yb atoms. YbC6, a famous kind of GICs, presents an AαAβ stacking where A corresponds to C layers and α and β represent the Yb intercalant layers. Interestingly, the lattice constant c significantly increases when Hubbard U increases; by contrast, the lattice constant a seems unaffected to Hubbard U and is approximately equals to 4.340 Å, which is close to the experimental value of 4.320 Å6,7 and the theoretical value of 4.340 Å.38 In fact, the lattice parameters a slightly differs in other GICs, such as 4.29 Å of NdC6, and 4.314 Å of EuC6.6,7
3.2 Elastic constants and polycrystalline moduli
The elastic constants of a solid link the mechanical and dynamical behaviors and provide important information regarding the nature of the forces operating in the solids, such as stability and stiffness. The independent elastic constants of YbC, Yb2C3, YbC2 and YbC6 with various Hubbard U values are calculated from the strained structures, and the relationship between independent elastic constants and Hubbard U values is plotted in Fig. 3. There are three independent elastic constants for cubic YbC and Yb2C3, i.e., C11, C12, and C44. The elastic constants of YbC and Yb2C3 decrease as Hubbard U increases except C12 of YbC. The elastic constant C12 of YbC first falls to 40.1 GPa at Hubbard U = 3 eV and then rises to 50 GPa at Hubbard U = 5 eV, forming a valley at Hubbard U = 3 eV, as shown in Fig. 3(a). For the relatively low-symmetry tetragonal YbC2 and hexagonal YbC6, there are six independent elastic constants. Similar to the elastic constants of YbC and Yb2C3 systems, most of the elastic constants of YbC2 and YbC6 decrease when Hubbard U increases. The elastic constant C33 of YbC2 and YbC6 decreases by approximately 23% and 14% from Hubbard U = 0 eV to 5 eV, respectively. Interestingly, the elastic constant C13 of YbC2 and YbC6 initially decreases and then increases as Hubbard U increases. The elastic constant C11 of YbC6 is the largest among the elastic constants of the four ytterbium carbides; in particular, C11 of YbC6 is 719 GPa and 713 GPa at Hubbard U = 0 and 5 eV, respectively. Moreover, the elastic constant C66 of YbC6 remains unchanged at 314 GPa with Hubbard U, as shown in Fig. 3(d). When the independent elastic constants are obtained, the intrinsic mechanical stability of the structure at various Hubbard U = 0, 1, 3 and 5 eV can be verified by the Born–Huang lattice dynamical theory. The criteria to determine the mechanical stability of cubic, tetragonal and hexagonal crystals are depicted as follows:39
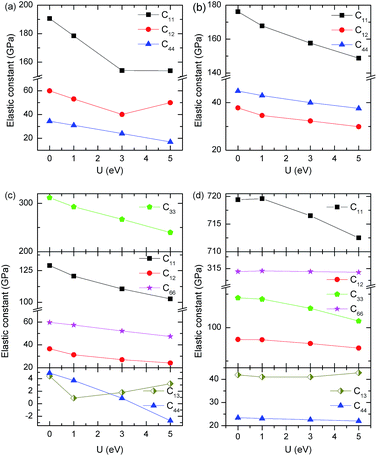 |
| | Fig. 3 Dependence of the elastic constants on Hubbard U for (a) YbC, (b) Yb2C3, (c) YbC2, and (d) YbC6. | |
Cubic phase (for YbC and Yb2C3):
| C11 − C12 > 0, C11 + 2C12 > 0, C44 > 0. |
Tetragonal phase (for YbC2):
| C11 > |C12|, (C11 + 2C12)C33 > 2C132, C44 > 0, C66 > 0. |
Hexagonal phase (for YbC6):
| C11 > |C12|, (C11 + 2C12)C33 > 2C132, C44 > 0. |
After these criteria are verified, the four ytterbium carbides with various Hubbard U values satisfy the criteria of mechanical stability except tetragonal YbC2 with Hubbard U = 5 eV where C44 is negative. Specially, it is worth noticing that the NaCl-type YbC is mechanically stable, although this carbide has not been synthesized successfully.
Mechanical properties are mainly determined by the quantity including bulk modulus B, Young's modulus E, shear modulus G, Pugh's ratio B/G and Poisson's ratio ν. Among these mechanical quantities, B and G can be obtained by Voigt–Reuss–Hill (VRH) approximation.40–42 Under the VRH BV, BR, GV, and GR, where the subscripts V and R represent Voigt and Reuss estimations of B and G, respectively, in different crystal systems can be expressed as follows:
Cubic phase (for YbC and Yb2C3):43,44
| BV = BR = (C11 + 2C12)/3, |
| GV = (C11 − C12 + 3C44)/5, |
| GR = 5(C11 − C12)C44/[4C44 + 3(C11 − C12)]. |
Tetragonal phase (for YbC2):45
| BV = (1/9)[2(C11 + C12) + 4C13 + C33], |
| GV = (1/30)(M + 3C11 − 3C12 + 12C44 + 6C66), |
| GR = 15[18BV/C2 + 6/(C11 − C12) + 6/C44 + 3/C66]−1, |
| M = C11 + C12 + 2C33 − 4C13, |
| C2 = (C11 + C12)C33 − 2C132. |
Hexagonal phase (for YbC6):46
| BV = (1/9)[2(C11 + C12) + 4C13 + C33], |
| GV = (1/30)(M + 12C44 + 12C66), |
| GR = (5/2)[C2C44C66]/[3BVC44C66 + C2(C44 + C66)], |
| M = C11 + C12 + 2C33 − 4C13, |
| C2 = (C11 + C12)C33 − 2C132. |
Hence, B and G are given as follows:
| B = 1/2(BR + BV) and G = 1/2(GR + GV). |
Once B and G are obtained, E and υ can be defined as follows:
| E = 9BG/(3B + G) and υ = (3B – 2G)/[2(3B + G)]. |
The dependence of BV, BR, GV, and GR on Hubbard U of YbC, Yb2C3, YbC2, and YbC6 is shown in Fig. 4. BV, BR, GV, and GR likely decrease as Hubbard U values increases except BV and BR of YbC. BV and BR of YbC first decreases from 103.5 GPa at U = 0 eV to 78.1 GPa at U = 3 eV and then increases to 84.7 GPa at U = 5 eV in accordance with the relationship of C11 and C12 as Hubbard U. By contrast, Hubbard U slightly affect B and G of YbC2 and YbC6, and this result is consistent with the variation in elastic constants.
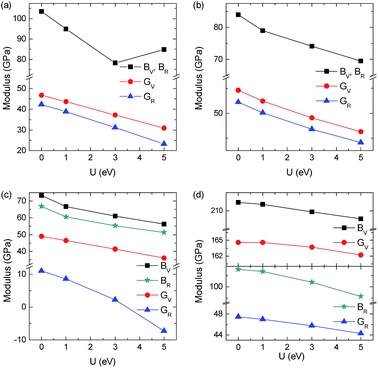 |
| | Fig. 4 Calculated bulk modulus B and shear modulus G of YbC, Yb2C3, YbC2 and YbC6 under Voigt and Reuss approximations with various Hubbard U values. | |
Bulk modulus B is measure of the resistance of a solid to volume change; shear modulus G represents the resistance to plastic deformation; and Young's modulus E denotes the resistance to uniaxial deformation. The calculated B, G, and E of the four ytterbium carbides with Hubbard U = 3 eV are listed in Table 1. Among the four ytterbium carbides, YbC6 yields the largest B of 155.9 GPa, G of 104.8 GPa, and E of 256.7 GPa. This result indicates that YbC6 is stiffer than the three other ytterbium carbides. On the contrary, YbC2 exhibits the smallest B of 58.2 GPa, G of 21.7 GPa, and E of 58.0 GPa. B of YbC2 is only approximately one-third of that of YbC6. Hardness and ductile will not coexist in a material. The ductile or brittle properties of materials can be related empirically to Pugh's ratio B/G. If B/G > 1.75, materials are ductile; otherwise, materials are brittle.47 The smallest B/G of 1.49 observed in YbC6 indicates brittleness. The largest B/G of 2.68 is detected in YbC2; this finding shows that YbC2 is relatively soft and ductile. Poisson's ratio ν generally quantifies the stability of a solid against shear deformation. For ductile materials, ν is approximately 0.33.48 The ductility of YbC2 is confirmed by the calculated ν of 0.33. Interestingly, B and G of YbC and Yb2C3 yield the same order of magnitude; however, B/G of YbC and Yb2C3 is more or less 1.75; thus, these carbides exhibit different degrees of brittleness.
Table 1 Bulk modulus B, shear modulus G, Young's modulus E, Pugh's ratio B/G, and Poisson ratio ν calculated with VRH approximations of YbC, Yb2C3, YbC2 and YbC6 at Hubbard U = 3 eV
| |
B (GPa) |
G (GPa) |
E (GPa) |
B/G |
ν |
| YbC |
78.1 |
34.2 |
89.6 |
2.28 |
0.309 |
| Yb2C3 |
74.1 |
47.9 |
118.2 |
1.55 |
0.240 |
| YbC2 |
58.2 |
21.7 |
58.0 |
2.68 |
0.334 |
| YbC6 |
155.9 |
104.8 |
256.7 |
1.49 |
0.226 |
3.3 Elastic anisotropy
Anisotropic behaviors should be estimated on the basis of fundamental and technological aspects because known crystals are elastically anisotropic. Several indexes, including the percent anisotropy (AB and AG) and shear anisotropic factors (A1, A2, and A3) have been developed to evaluate elastic anisotropy.49 The percent anisotropy in compressibility and shear are defined as follows:
and
| AG = (GV − GR)/(GV + GR), |
respectively. For a completely isotropic system, AB and AG are 0, and the deviation from 0 measures the degree of elastic anisotropy. As shown in Table 2, the percent anisotropies in the shear AG of high-symmetry cubic YbC and Yb2C3 systems are 0.0877 and 0.0240, respectively; whereas AB equals 0 because of the same BV and BG. YbC6 possesses the largest AB of 0.3455 and a moderate AG of 0.5268, whereas YbC2 yields the largest AG of 0.8989. This result indicates that YbC2 displays a large anisotropy in shear. It is should be noted that AG of the four ytterbium carbides is larger than AB, suggesting that these carbides are slightly anisotropic in compressibility.
Table 2 Percent anisotropy (AB and AG) and shear anisotropic factors (A1, A2, and A3) of YbC, Yb2C3, YbC2, and YbC6 at Hubbard U = 3 eV
| |
AB |
AG |
A1 |
A2 |
A3 |
| YbC |
0 |
0.0877 |
0.4211 |
0.4211 |
0.4211 |
| Yb2C3 |
0 |
0.0240 |
0.6385 |
0.6385 |
0.6385 |
| YbC2 |
0.0490 |
0.8989 |
0.0095 |
0.0095 |
1.2488 |
| YbC6 |
0.3455 |
0.5628 |
0.1207 |
0.1207 |
0.9997 |
Shear anisotropic factors correspond to the degree of anisotropy in the bonding between atoms in different planes. A1 of the (100) shear planes between [011] and [010] directions, A2 of the (010) shear planes between [101] and [001] directions, and A3 of the (001) shear planes between [110] and [010] directions are defined as follows:
| A1 = (4C44)/(C11 + C33 − 2C13), |
| A2 = (4C55)/(C22 + C33 − 2C23), |
and
| A3 = (4C66)/(C11 + C22 − 2C12). |
The three indexes must be identical for a completely isotropic system, and any deviation from unity corresponds to the degree of shear anisotropy. A1, A2, and A3 are identical in YbC and Yb2C3 because of the shear isotropy of cubic structure. A1 of YbC2 is smaller than that of YbC6, indicating that YbC2 behaves more (100) shear anisotropically than YbC6. A3 of 0.9997 of YbC6 demonstrates the almost isotropic property of (001) shear planes between [110] and [100] directions.
A three-dimensional surface construction of B and E, along with their projections on specific planes, is plotted to describe the anisotropy of the mechanical moduli of the four ytterbium carbides. The reciprocal of B and E are defined as follows:50
Cubic phase (for YbC and Yb2C3):
Tetragonal phase (for YbC2):
Hexagonal phase (for YbC6):
where
l1,
l2, and
l3 are the direction cosines in various systems, and
sij is elastic compliance constant obtained using the GGA +
U scheme with Hubbard
U = 3 eV.
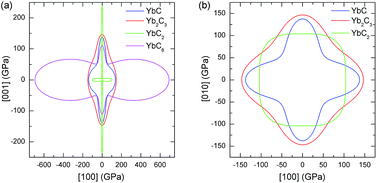
The surface constructions of B of YbC, Yb2C3, YbC2, and YbC6 are displayed in Fig. 5, and their B projections on the (010) and (001) planes are plotted in Fig. 6. The surface constructions of B of YbC and Yb2C3 exhibit a perfect sphere, indicating isotropic characteristics because of high-symmetry cubic lattice, see Fig. 5(a) and (b). B of tetragonal YbC2 and hexagonal YbC6 shows strong anisotropic features. The surface construction of B of YbC2 is likely an ellipsoid, see Fig. 5(c), whereas the surface of B of YbC6 is similar to a disc, see Fig. 5(d). It is worth noticing that these results are in good agreement with the calculated anisotropic factors and indexes listed in Table 2. B projections on the (010) and (001) planes provide further details regarding anisotropic properties. The x axis range is five times larger than the y axis range to display B projections of the four carbides. The B projection on the (010) plane of YbC and Yb2C3, resembling an ellipse in Fig. 6(a), should be a circle. The B projection on the (010) plane of YbC6 exhibits a butterfly shape, showing a strong anisotropic property. B projected on the (001) plane of the four ytterbium carbides is circular, which indicates an isotropic property. Due to the large B of YbC6, the circular B on the (001) plane is noticeably larger than the three other ytterbium carbides. Fig. 6(b) illustrates only B of YbC, Yb2C3, and YbC2 but excludes B of YbC6 to show the B projection. The circular area of YbC is slightly larger than that of Yb2C3, which is consistent with B listed in Table 1.
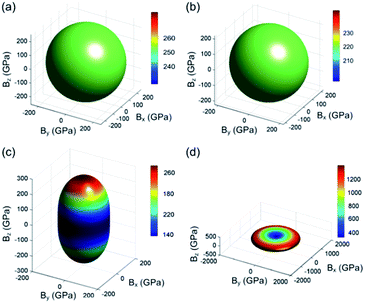 |
| | Fig. 5 Surface constructions of bulk modulus B of (a) YbC, (b) Yb2C3, (c) YbC2, and (d) YbC6. | |
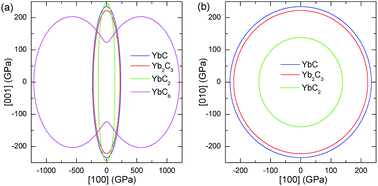 |
| | Fig. 6 Bulk modulus B projections on (a) (010) and (b) (001) plane of YbC, Yb2C3, YbC2, and YbC6. | |
The surface constructions of E of YbC, Yb2C3, YbC2, and YbC6 are displayed in Fig. 7, and E projections on the (010) and (001) planes are plotted in Fig. 8. The anisotropic property of E of YbC, Yb2C3, YbC2, and YbC6 is more evident than that of B. The surface constructions of E of YbC and Yb2C3 resemble a similar shape, see Fig. 7(a) and (b). E along the [100], [010], and [001] directions are considerably larger than E along other directions, indicating anisotropic features. From Fig. 8(b), it can be seen that E of YbC projection on the (001) plane is smaller than that of Yb2C3, which is consistent with that of VRH approximations listed in Table 1. The surface construction of E of YbC2 is similar to a fishing float with a large value along the [001] direction, showing a strong anisotropic property. The projection of E of YbC2 exhibits a rounded square, as seen in Fig. 8(b). Similar to the surface construction of B, the surface construction of E of YbC6 is similar to a disc perpendicular to the [001] direction. Interestingly, E of YbC6 in the [100] direction is obviously larger than that in the [001] direction, as shown in Fig. 8(a). The projection E on the (001) plane of YbC6 is a circle but is not shown in Fig. 8(b) because of its large area.
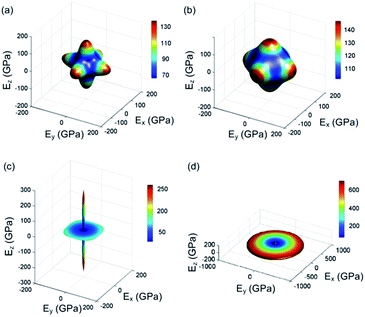 |
| | Fig. 7 Surface constructions of Young's modulus E of (a) YbC, (b) Yb2C3, (c) YbC2, and (d) YbC6. | |
 |
| | Fig. 8 Young's modulus E projections on (a) (010) and (b) (001) plane of YbC, Yb2C3, YbC2, and YbC6. | |
3.4 Anisotropy in acoustic velocities
The phase velocities of pure transverse and longitudinal modes of YbC, Yb2C3, YbC2, and YbC6 are investigated from single crystal elastic constants in accordance with the procedure developed by Brugger.51,52 The symmetry of crystal indicates that pure transverse and longitudinal modes can exist along specific directions: [001], [110], and [111] for a cubic crystal; [001], [110], and [100] for a tetragonal crystal; and [001] and [110] for a hexagonal crystal. In each direction, two transverse modes and one longitudinal mode are involved.53 The calculated sound velocities of ytterbium carbides with Hubbard U = 3 eV are shown in Table 3.
Table 3 Sound velocities (km s−1) along different directions for YbC, Yb2C3, YbC2 and YbC6 with Hubbard U = 3 eV
| |
|
YbC |
Yb2C3 |
YbC2 |
YbC6 |
| [001] |
[001]vl |
4.019 |
4.245 |
6.101 |
4.563 |
| [110]vt1 |
1.586 |
2.139 |
0.354 |
2.028 |
| [110] |
[110]vl |
3.847 |
4.157 |
4.108 |
11.419 |
[![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10]vt1 10]vt1 |
2.445 |
2.677 |
2.414 |
7.562 |
| [001]vt2 |
1.586 |
2.139 |
0.354 |
2.028 |
| [111] |
[111]vl |
3.397 |
3.817 |
|
|
[11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ]vt1 ]vt1 |
2.196 |
2.510 |
|
|
| [100] |
[100]vl |
|
|
3.927 |
|
| [010]vt1 |
|
|
2.698 |
|
| [001]vt2 |
|
|
0.354 |
|
In the two cubic phases, the sound velocities of Yb2C3 are larger than those of YbC in the [001], [110], and [111] directions. For example, the longitudinal and transverse mode velocities of Yb2C3 in the [001] direction are 5.6% and 34.8% larger than those of YbC. The longitudinal and transversemode velocities of the cubic system are proportional to C11 and C44, respectively, and are inversely proportional to the density ρ which is dominated by carbon contents. C11 and C44 of Yb2C3 are larger than those of YbC, and the density ρ of Yb2C3 is smaller than that of YbC due to the relatively higher carbon contents. This leads to that the average sound velocities of Yb2C3 are larger than that of YbC, as shown in Fig. 9. The longitudinal and transverse vibration modes in the [001] direction of YbC2 are parallel and perpendicular to the C2 dimer stretching mode. Thus, YbC2 yields a relatively larger longitudinal mode velocity of 6.10 km s−1 and an extremely low transverse mode velocity of 0.35 km s−1. Moreover, the significant difference between the transverse and longitudinal velocities in the [001], [100], and [110] directions results in relatively low average velocities, as shown in Fig. 9.
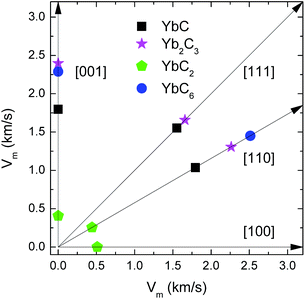 |
| | Fig. 9 Averaged sound velocities (vm) in the [100], [001], [110] and [111] directions of YbC, Yb2C3, YbC2, and YbC6. | |
The longitudinal mode velocity in the [110] direction of YbC6 is as high as 11.42 km s−1, whereas two transverse mode velocities along [10] and [001] directions are 7.562 km s−1 and 2.028 km s−1, respectively. The sound velocity is well correlated with the structural characteristics. The longitudinal mode [110]vl direction is the nearest C–C connecting direction in the six-membered C ring; conversely, the transverse mode [10]vt1 direction is the second-nearest C–C connecting direction in the six-membered C ring. Furthermore, the large longitudinal mode velocity in the [110] direction corresponds the highest average velocity along the [110] direction among the four ytterbium carbides.
3.5 Thermodynamic properties
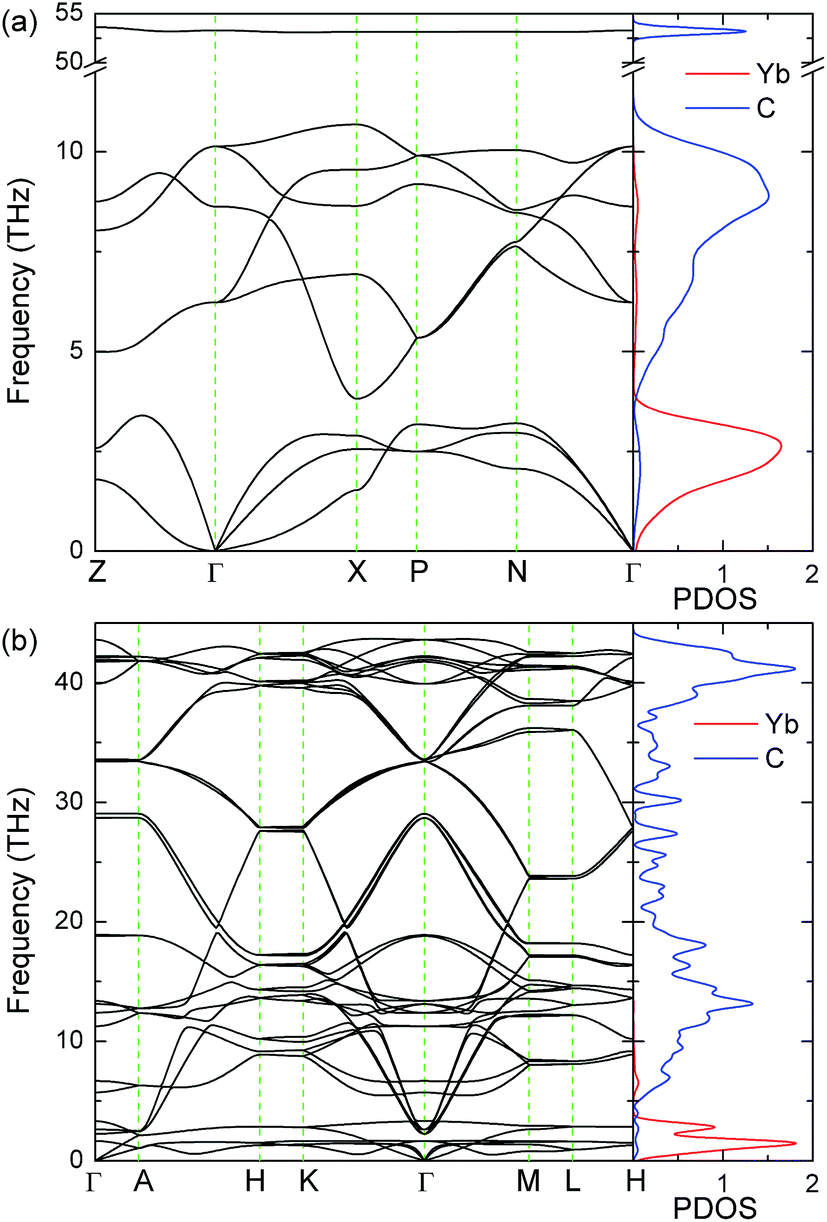
The calculated phonon band structures along some high-symmetry directions in Brillouin zone and the phonon projected density of states (PDOS) of YbC2 and YbC6 at Hubbard U = 3 eV are displayed in Fig. 10. Phonon calculations established the dynamical stability of YbC2 and YbC6 in view of the absence of imaginary frequencies. Additional phonon calculations of YbC and Yb2C3 with various Hubbard U parameters and sizes of supercells are performed. The results show that imaginary frequencies exist in their phonon band structures (data not shown here). This finding reveals that YbC and Yb2C3 are not thermodynamically stable, which does not coincide with the mechanical stability. This phenomenon is also found in UO2. The Pnma phase of UO2 is predicted to be mechanically stable, but is found to be thermodynamically unstable from phonon calculations.54
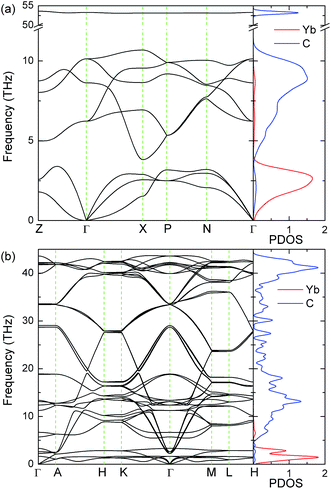 |
| | Fig. 10 Phonon band structures and PDOS of (a) YbC2 and (b) YbC6 with Hubbard U = 3 eV. The unit of PhDOS is states/THz/fu. | |
The vibration frequency of the Yb atom is apparently lower than that of the C atom because the Yb atom is considerably heavier than C atom. Thus, the phonon structures of both YbC2 and YbC6 can be divided into two major regions. The heavy Yb atoms dominate low-frequency modes below 4 THz, whereas the light C atoms contribute significantly to high-frequency vibrations. Compared with YbC6, the high-frequency region of YbC2 shifts upward and reaches approximately 53 THz. It is noteworthy that the flat regions of the phonon dispersion curves of YbC2 as shown in Fig. 10(a), which correspond to the peaks in the phonon PDOS, indicate the localization of the states, i.e., the high-frequency stretching of C2 units.
The temperature variations of Helmholtz free energy, entropy, and heat capacity at constant volume of YbC2 and YbC6 are shown in Fig. 11. It is noteworthy that the Helmholtz free energy at 0 K does not vanish because of zero-point motion. Helmholtz free energies per fu of YbC2 and YbC6 are 20.1 and 90.7 kJ mol−1, respectively. As temperature increases, Helmholtz free energies of YbC2 and YbC6 decrease; however, Helmholtz free energy of YbC6 rapidly decreases, leading to that it intersects with the Helmholtz free energy of YbC2 at 1350 K. The entropy of YbC6 is larger than that of YbC2, and the difference between the entropies of these carbides increases as temperature increases. The heat capacity at constant volume of YbC6 and YbC2 is nearly the same below 200 K. At an intermediate temperature range, CV is governed by atomic vibrations. Above 200 K, CV of YbC6 is larger than that of YbC2. At high temperature, CV of YbC2 becomes constant at 75 J mol−1 K, while CV of YbC6 reaches 170 J mol−1 K−1.
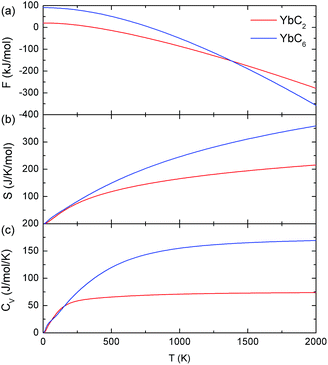 |
| | Fig. 11 Temperature dependence of (a) Helmholtz free energy (F), (b) entropy (S), and (c) heat capacity at constant volume (CV) of YbC2 and YbC6 with Hubbard U = 3 eV. | |
4. Conclusions
The mechanical and thermodynamic properties of YbC, Yb2C3, YbC2, and YbC6 have been investigated with the GGA + U frameworks. The calculated lattice constants of YbC, Yb2C3, YbC2, and YbC6 are found to increase as Hubbard U values increase. The four ytterbium carbides with various Hubbard U values satisfy the Born–Huang lattice dynamical criteria of mechanical stability except tetragonal YbC2 with Hubbard U = 5 eV. YbC6 is stiffer than the three other ytterbium carbides, whereas YbC2 is softest and most ductile among the four ytterbium carbides. The directional E and B, and shear anisotropic factors indicate the mechanical anisotropic properties of YbC, Yb2C3, YbC2 and YbC6. The phonon calculation reveals that YbC2 and YbC6 are thermodynamically stable, but NaCl-type YbC and Pu2C3-type Yb2C3 are thermodynamically unstable. These theoretical results are consistent with experimental findings.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 11304269 and 11304268).
References
- K. A. Gschneider, Rare earth carbides, 1991 Search PubMed.
- M. S. Sselhaus and G. Sselhaus, Adv. Phys., 2002, 51, 1–186 CrossRef.
- S. Ma, K. Bao, Q. Tao, X. Huang, P. Zhu and T. Cui, RSC Adv., 2014, 4, 63544–63548 RSC.
- D. D. Kumar, N. Kumar, S. Kalaiselvam, R. Radhika, S. Dash, A. K. Tyagi and R. Jayavel, RSC Adv., 2015, 5, 81790–81801 RSC.
- L. Brewer and O. Krikorian, J. Electrochem. Soc., 1955, 103, 701–703 CrossRef.
- M. E. Makrini, D. Guérard, P. Lagrange and A. Hérold, Physica B+C, 1980, 99, 481–485 CrossRef.
- M. E. Makrini, D. Guérard, P. Lagrange and A. Hérold, Carbon, 1980, 18, 203–209 CrossRef.
- M. H. Upton, T. R. Forrest, A. C. Walters, C. A. Howard, M. Ellerby, A. H. Said and D. F. McMorrow, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 134515 CrossRef.
- V. Babizhetskyy, O. Jepsen, R. K. Kremer, A. Simon, B. Ouladdiaf and A. Stolovits, J. Phys.: Condens. Matter, 2014, 26, 025701 CrossRef CAS PubMed.
- C. Zhang, S. T. John, T. Kaori and H. Q. Lin, EPL, 2012, 100, 67003 CrossRef.
- Y. L. Li, W. Luo, Z. Zeng, H. Q. Lin, H. K. Mao and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9289–9294 CrossRef CAS PubMed.
- Y. L. Li, S. N. Wang, A. R. Oganov, H. Gou, J. S. Smith and T. A. Strobel, Proc. Natl. Acad. Sci. U. S. A., 2015, 6, 6974 CAS.
- Y. L. Li, W. Luo, X. J. Chen, Z. Zeng, H. Q. Lin and R. Ahuja, Sci. Rep., 2013, 3, 3331 Search PubMed.
- F. H. Spedding, K. Gschneidner and A. H. Daane, J. Am. Chem. Soc., 1958, 80, 4499–4503 CrossRef CAS.
- J. M. Haschke and H. A. Eick, J. Am. Chem. Soc., 1970, 92, 1526–1530 CrossRef CAS.
- K. A. Gschneidner and F. W. Calderwood, Bull. Alloy Phase Diagrams, 1986, 7, 568–570 CrossRef CAS.
- M. Atoji and R. H. Flowers, J. Chem. Phys., 1970, 52, 6430–6431 CrossRef CAS.
- T. E. Weller, M. Ellerby, S. S. Saxena, R. P. Smith and N. T. Skipper, Nat. Phys., 2005, 1, 39–41 CrossRef CAS.
- G. Csányi, P. B. Littlewood, A. H. Nevidomskyy, C. J. Pickard and B. D. Simons, Nat. Phys., 2005, 10, 1038 Search PubMed.
- I. I. Mazin, Phys. Rev. Lett., 2005, 95, 227001 CrossRef CAS PubMed.
- M. Calandra and F. Mauri, Phys. Rev. Lett., 2005, 95, 237002 CrossRef PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys.
Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS.
- I. I. Mazin and S. L. Molodtsov, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 172504 CrossRef.
- M. Neupane, S. Y. Xu, N. Alidoust, G. Bian, D. J. Kim, C. Liu, I. Belopolski, T. R. Chang, H. T. Jeng and T. Durakiewicz, Phys. Rev. Lett., 2015, 114, 016403 CrossRef PubMed.
- G. Kresse and J. Furthmüler, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- A. Togo, L. Chaput, I. Tanaka and G. Hug, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174301 CrossRef.
- T. Mochiku, T. Nakane, H. Kito, H. Takeya, S. Harjo, T. Ishigaki, T. Kamiyama, T. Wada and K. Hirata, Phys. C, 2005, 426–431, 421–425 CrossRef CAS.
- R. K. Kremer, J. S. Kim, W. H. Xie, V. Babizhetskyy, O. Jepsen, A. Simon, K. S. Ahn, B. Raquet, H. Rakoto and J. M. Broto, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 014516 CrossRef.
- D. W. Jones, I. J. McColm, R. Steadman and J. Yerkess, J. Solid State Chem., 1984, 53, 376–381 CrossRef CAS.
- M. Atoji, J. Chem. Phys., 1961, 35, 1950–1960 CrossRef CAS.
- C. M. Fang, J. Bauer, J. Y. Saillard and J. F. Halet, Z. Naturforsch., 2007, 38, 971–976 Search PubMed.
- F. Mouhat and F. X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- W. Voigt, Lehrburch der Kristallphysik, Teubner, Leipzig, 1928 Search PubMed.
- A. Reuss, Z. Angew. Math. Mech., 1929, 9, 49 CrossRef CAS.
- R. Hill, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–354 CrossRef.
- J. Feng, B. Xiao, R. Zhou, W. Pan and D. R. Clarke, Acta Mater., 2012, 60, 3380–3392 CrossRef CAS.
- X. Gao, Y. Jiang, R. Zhou and J. Feng, J. Alloys Compd., 2014, 587, 819–826 CrossRef CAS.
- J. P. Watt, J. Appl. Phys., 1986, 60, 3120–3124 CrossRef CAS.
- J. P. Watt and L. Peselnick, J. Appl. Phys., 1980, 51, 1525–1531 CrossRef CAS.
- S. F. Pough, Philos. Mag., 1954, 45, 823–843 CrossRef.
- J. Haines, A. Jm Léger and G. Bocquillon, Annu. Rev. Mater. Res., 2001, 31, 1–23 CrossRef CAS.
- P. Ravindran, L. Fast, P. A. Korzhavyi and B. Johansson, J. Appl. Phys., 1998, 84, 4891–4904 CrossRef CAS.
- J. F. Nye, Physical properties of crystals, Oxford University Press, Oxford, UK, 1985 Search PubMed.
- K. Brugger, J. Appl. Phys., 1965, 36, 768–773 CrossRef.
- K. Brugger, J. Appl. Phys., 1965, 36, 759–768 CrossRef.
- J. Feng, B. Xiao, Z. C. Huang, J. C. Chen, C. L. Wan, Z. X. Qu, R. Zhou and W. Pan, Acta Mater., 2010, 72, 1742–1760 Search PubMed.
- B. T. Wang, P. Zhang, R. Lizárraga, I. Di Marco and O. Eriksson, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 1047 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.