
An ultrasensitive electrochemical immunosensor for the detection of human immunoglobulin G based on Ag@BSA microspheres†
Hongfang Zhang*a,
Danlei Ninga and
Jianbin Zheng*b
aMinistry of Education Key Laboratory of Synthetic and Natural Functional Molecular Chemistry, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, P. R. China. E-mail: zhanghf@nwu.edu.cn
bShaanxi Provincial Key Laboratory of Electroanalytical Chemistry, Northwest University, Xi'an 710069, P. R. China. E-mail: zhengjb@nwu.edu.cn
First published on 9th December 2015
Abstract
A novel human immunoglobulin G (HIgG) electrochemical immunosensor was developed based on nanosilver-doped bovine serum albumin microspheres (Ag@BSA). The immunosensor was prepared step-wise by first modifying the electrode with β-cyclodextrin functionalized gold nanoparticles followed by the immobilization of captured antibodies and then the formation of a sandwich-type immunocomplex to introduce Ag@BSA bionanoprobes on the sensor surface. The amplification pathway using the stripping voltammetric measurement of silver ions released from Ag@BSA to monitor the immunoreaction was first adopted. The immunosensor exhibited a large dynamic range of 1 fg mL−1 to 10 pg mL−1 and an ultralow detection limit of 0.5 fg mL−1 to HIgG. Moreover, the immunosensor also showed acceptable stability and reproducibility. This biosensor was applied to the detection of the HIgG level in real serum samples.
Introduction
The level of certain biomarkers, such as human immunoglobulin G (HIgG) in serum, can indicate a variety of disease characteristics and provide key information on the humoral immune status.1,2 The sensitive determination of important biomarkers is therefore of great significance in clinical research and disease diagnosis. An immunoassay based on the antigen–antibody specific recognition is one of the major analytical techniques in clinical diagnoses.3 Among various analytical tools, electrochemical immunosensors have drawn considerable interest not only due to their intrinsic advantages of low cost and simple instruments, but also because of their relatively low detection limit and formidably high sensitivity.4,5 The performance of the electrochemical immunosensor was greatly influenced by the electron transfer efficiency and biocompatibility of materials modified on the electrode surface.6,7 From this point of view, noble metal nanoparticles were frequently chosen for the sensing interface and tracing tags.3–6 However, it was a challenge to decrease the background signal produced in the absence of antigens which often restricted the detection limit of the immunosensor.8–10 The high background signal, in the vast majority of cases, was caused by the nonspecific adsorption of electroactive labels on the sensing interface.7,10,11Bioinspired nanomaterial such as the protein-directed inorganic nanoparticles is a rising-star of nanomaterial. The protein can help to prevent aggregation of nanoparticles and allow the materials to be chemically modified for targeting. The inorganic nanoparticles can exhibit dramatically different structural and electronic properties with the materials synthesized without protein templating. Among them, bovine serum albumin (BSA)-mediated nanomaterial has attracted increasing attention due to green reaction process and multi-functionality of the products.12–19 BSA-stabilized Pt, Au and Ag nanoclusters have been successfully explored to fabricate sensing interfaces.14–18 These nanoclusters provided with some advantageous properties, but not limited to, favorable biocompatibility, excellent conductivity, and enhanced electrocatalytic activity toward oxygen or H2O2 reduction.15 Using Au@BSA or Ag@BSA microspheres as the biomimetic sensing layer, Jia's distinguished group13,16,17 developed electrochemical biosensor for urinary retinal-binding protein, carcinoembryonic antigen-positive cells and KB cells, respectively. Zhou et al.12 devised a new sandwich-type electrochemical immunoassay for the detection of carcinoembryonic antigen. The results indicated that the labeled antibody on the Ag@BSA could maintain the native bioactivity for the antigen–antibody reaction. Ma et al.19 expanded the application scope of BSA-stabilized nanomaterials by a 3D origami electrochemical immunodevice. They select Au@BSA nanospheres as nanocarriers for loading numerous metal ions such as Pb2+ and Cd2+ to form Au@BSA–metal ion tracers. Since nanosilver itself can be electrochemically oxidized at low potential and yield a well-shaped stripping peak,5,8 Ag@BSA can be adopted as desirable labels for electrochemical immunoassay.
In this work, Ag@BSA microspheres were applied as the nanocarriers for antibodies, electroactive labels for detection and nanoprobes for signal amplification. As illustrated in Scheme 1, the primary antibodies were immobilized onto the electrode surface via the adsorption and special inclusion ability of β-cyclodextrin functionalized gold nanocomposite (β-CD/Au).20–24 Then, Ag@BSA nanocomposites were introduced on the immunosensor surface by the formation of sandwich-type immunocomplex. Dissolution of the silver on the sensor surface gave rise to a “burst” of Ag+ ions which could produce sensitive peak on the anodic stripping voltammogram. To the best of our knowledge, this is the initial attempt to monitor immunoreaction by anodic stripping voltammetric (ASV) determination of silver ions released from Ag@BSA. With HIgG as a model analyte, the fabricated immunosensor exhibited a low detection limit of 0.5 fg mL−1.
 | ||
| Scheme 1 Illustration for preparation of β-CD/Au (A), nanolabels (B) and the immunosensor. | ||
Experimental
Materials and reagents
HIgG, mouse anti-HIgG antibody (Ab) and BSA were obtained from Beijing Boisynthesis Biotechnology Co. Ltd. (Beijng, China). Glutaraldehyde (GA) was purchased from Kemiou Chemical Reagent Co. Ltd. (Tianjin, China). β-CD, trisodium citrate and silver nitrate (AgNO3) were obtained from Shanghai Reagent Company (Shanghai, China). Sodium borohydride (NaBH4) and nitric acid (HNO3) were obtained from Sinopharm Chemical Reagent Co. Ltd. (China). Tween-20 was obtained from MP Biomedicals. All other reagents were of analytical grade and used as received.Phosphate buffered saline (PBS) was prepared with Na2HPO4·12H2O and KH2PO4. PBS (0.01 M, pH 7.4) containing 1.0% (w/v) BSA and PBS containing 0.05% (V/V) Tween-20 (PBST) was used as blocking and washing solution, respectively. Deionized and distilled water was used throughout the study.
Apparatus
All electrochemical experiments were achieved on a CHI660A electrochemical workstation (Shanghai CH Instrument Co. Ltd., China) using a three-electrode system. The working electrode was glassy carbon electrode (GCE) or modified GCE. A saturated calomel electrode (SCE) and a platinum wire served as reference and counter electrode, respectively. UV-vis analysis was performed on UV-2550 spectrophotometer (Shimadzu, Japan). Scanning electron microscopy (SEM) images were obtained using a JSM-6390A (JEOL, Japan). FTIR spectrum was conducted on TENSOR-27 FTIR spectrometer (Bruker, GER). The transmission electron microscopy (TEM) images were obtained from Tecnai G2 F20 S-TWIN (FEI, USA).Synthesis of β-CD functionalized Au nanoparticles
In a typical procedure, 1.0 mL of 0.01 M β-CD was mixed with 0.1 mL of 0.01 M HAuCl4 in 3.9 mL of deionized water at room temperature. 0.01 mL of 1.0 M NaOH was injected into the mixture with stirring to adjust to the pH 10.5 of a solution. After being vigorously stirred, the mixture was continuously heated at 60 °C for 3 hours in a water bath. The color of the solution was transferred from pink to wine red, indicating to the formation of AuNPs. UV-vis spectrum of β-CD/Au (Fig. S1†) was in accordance with a previous report.24Synthesis of Ag@BSA microspheres
Ag@BSA microspheres were synthesized using a slightly modified recipe by Jia's group.13 Initially, at room temperature, 5 mL of 0.05 M AgNO3 solution was added to 10 mL of 3.0 mg mL−1 BSA solution under stirring, and then the mixture kept static under N2 for 5 hours. Afterward, 0.2 mL of hydrazine monohydrate solution was rapidly injected into the mixture and continuously stirred. After being stirred for 30 min, the mixture was aged at room temperature for 48 h. Then, the resulting solution was centrifuged at 5000 rpm for 10 min. Finally, the obtained product was re-dispersed in deionized water and stored at 4 °C before use.Conjugation of Ag@BSA microspheres with detection antibody
The conjugation of detection antibody (Ab2) on Ag@BSA microspheres were prepared by this protocol. 600 μL of GA solution (excess) was dispersed into 4.0 mL of Ag@BSA suspension (1.0 mg mL−1) under vigorous stirring, following by incubation at room temperature overnight. The excess GA was removed by centrifugation and the obtained sample was redispersed in 2.0 mL of 0.5 M Na2CO3. Afterwards, 100 μL of Ab2 (100 μg mL−1) were injected into the mixture and stirred for 10 min, the mixture was transferred to the refrigerator at 4 °C for further reaction (overnight). During this process, antibodies were covalently conjugated onto the surface of Ag@BSA microspheres. Finally, the suspension was separated by centrifugation (8000 rpm, 10 min) and redispersed in 4.0 mL of 0.01 M PBS (pH 7.4).Preparation of the electrochemical immunosensor
In order to prepare the modified electrode, 5 μL of β-CD/Au dispersion was coated on the GCE. After dried, 5 μL of 0.2 mg mL−1 anti-Human IgG (Ab1) was dropped on the surface of β-CD/Au/GCE, which was incubated at 4 °C overnight. The excess antibodies were removed with PBST and PBS. Finally, a drop of 5 μL blocking solution was covered on the electrode surface and incubated for 30 min at 37 °C to block possible remaining active sites against nonspecific adsorption. After another washing with PBST and PBS, the immunosensor was obtained and stored at 4 °C prior to use.Measurement procedure
To carry out the immunoreaction and electrochemical measurement, the immunosensor was incubated at 37 °C for 30 min with different concentration of HIgG. After washing, the immunosensor was further incubated with a certain volume of Ag@BSA-Ab2 for 40 min at 37 °C. After rinsing, the silver ions were released by adding 200 μL of 1.5 M HNO3 and quantified by ASV at following conditions: 10 min deposition at −0.5 V, and potential scan at the linear sweep voltammetry (LSV) from −0.3 to 0.3 V at 50 mV s−1 was performed in a cell containing 5.0 mL of 0.6 M KNO3 as the electrolyte solution to record the response.Results and discussion
Characterization of Ag@BSA microspheres
To study the pictorial presentation of the silver nanoparticles in solution, UV-vis spectrum of Ag@BSA was recorded (Fig. 1A). A broad peak at around 440 nm was observed, which is consistent with the previous reporting for the surface plasmon peak of Ag@BSA microspheres.25,26 The structure of the microspheres was further characterized by the techniques of SEM (Fig. 1B and C) and TEM (Fig. 1D). The uniform microspheres with a mean size of 500 nm in diameter and rough surface were acquired. The functional groups on the large surface of Ag@BSA make the microspheres appropriate for the covalent attachment of antibodies.16,17 | ||
| Fig. 1 UV-vis spectrum (A), SEM (B and C) and TEM (D) images of Ag@BSA. | ||
Characteristics of the immunosensor and signal amplification
The impedance change of the interfacial property of the electrode was characterized using electrochemical impedance spectroscopy (EIS). It is well known that the semicircle portion of Nyquist plot at high frequencies represents electron transfer-limited process, where the diameter equals the electron transfer resistance.4 As shown in Fig. 2A, bare GCE displayed an ultra-small semicircle (curve a), suggesting a characteristic of diffusion-limit step of the electrochemical process.27 After β-CD/Au was modified on the surface of the GCE (curve b), the semicircle diameter increased slightly compared with bare GCE, which demonstrated the excellent electronic conductivity of β-CD/Au. EIS of β-CD modified electrode was also recorded. The diameter of the semicircle (shown in curve c) was apparently bigger than that for β-CD/Au/GCE, demonstrating that the nanogold plays an important role in facilitating the electron exchange between the solution and the electrode. The diameter increased consecutively with the immobilization of Ab1 (curve d), the incubation of HIgG (curve e) and then Ab2-Ag@BSA (curve f), indicating that the immunocomplex had formed on the electrode surface and thus hindered the electron transfer.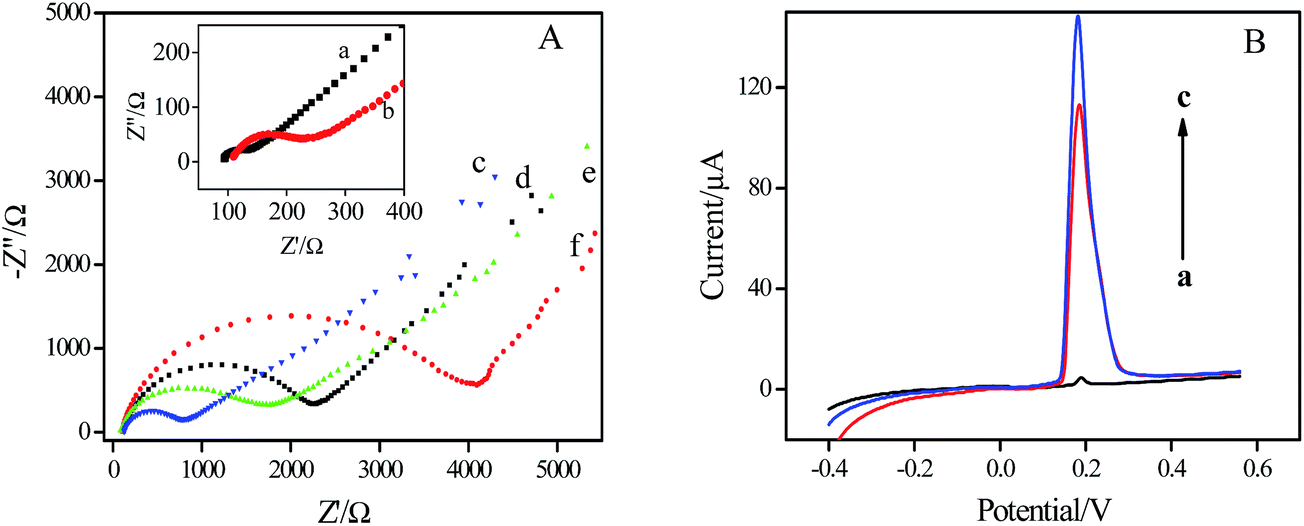 | ||
| Fig. 2 (A) EIS obtained for different modified electrodes in KCl solution containing 0.5 mM Fe(CN)63−/Fe(CN)64−. (a) GCE, (b) β-CD-Au/GCE, (c) β-CD/GCE, (d) Ab1/β-CD/Au/GCE, (e) HIgG/BSA/Ab1/β-CD-Au/GCE, (f) Ab2-Ag@BSA/HIgG/BSA/Ab1/β-CD/Au/GCE. (B) ASV of the immunosensor after sandwich immunoreactions with blank control (a), 10 pg mL−1 (b) and 500 pg mL−1 HIgG (c) in 0.6 M KNO3 solution. Deposition potential: −0.5 V; deposition time: 10 min; scan rate: 50 mV s−1. | ||
The release of Ag+ from Ag@BSA microspheres following the immunoassay sandwich formation was firstly adopted to quantitatively monitor the immunoreaction. Fig. 2B depicts the anodic stripping voltammetric curves of this immunosensor toward blank buffer and HIgG, respectively. As shown in curve a, the immunosensor incubated with blank buffer gave rise to a small peak which was produced by the nonspecific adsorption of Ag@BSA on the electrode surface. The peak current is only about 4.2 μA, which is negligible compared with the response for 10 pg mL−1 HIgG shown in curve b, Fig. 2B, where a well-defined stripping peak at 0.18 V, corresponds to the oxidation of Ag,23,28 was observed. The peak current increased when the concentration of target protein increased from 10 pg mL−1 to 500 pg mL−1 (curve c, Fig. 2B), indicating that the Ag@BSA microspheres-based strategy is appropriate for the quantitative detection of HIgG.
Optimization of experimental conditions
The preconcentration step is critical in any stripping technique, and thus we investigated the effect of sliver accumulation time on immunosensor response. As shown in Fig. 3A, the peak current on ASV of enriched silver increased linearly when the silver accumulation time was varied from 1 to 30 min. Considering both sensitivity and analytical time, we chose 10 min for the optimized silver enrichment. | ||
| Fig. 3 Dependence of stripping peak current of the immunosensor on (A) incubation time on the amount of sliver enrichment, (B) antibody concentration (C) incubation time for the antigen–antibody. | ||
The concentration of capture antibody was optimized. As shown in Fig. 3B, when the concentration of anti-HIgG changed from 0.05 to 0.5 mg mL−1, the peak current increased till it nearly reached a plateau at 0.2 mg mL−1. The possible reason for the trend was that more antigen-binding sites were accessible when more antibodies were immobilized. Higher concentration than 0.2 mg mL−1 of antibodies might increase disorder in alignment of adsorbed antibody and steric hindrance of immunoreaction.27 So, 0.2 mg mL−1 Ab1 was selected for the following experiments. The effect of the incubation time for HIgG and Ab2-Ag@BSA was also investigated. As shown in Fig. 3C, the ASV response of the immunosensor increased with an increment of incubation time and tended to level off at 40 min, which showed the saturated state of the sandwich immunoreactions. Therefore, the incubation time of 40 min was adopted as the optimal condition in this work.
Analytical performance
Under optimal conditions, ASV was used to investigate the relationship between the peak current values and HIgG concentration. The ASV response of the immunosensor for HIgG measurement increased with increasing the concentration of the analyte (Fig. 4A). The calibration plot showed two linear segments between the peak currents and the logarithm of analyte concentrations in the range from 1 fg mL−1 to 10 pg mL−1 (Fig. 4B). And the equations were I = 56.31 + 14.33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lgC (pg mL−1) with a correlation coefficient of 0.994 and I = 18.01 + 46.97lgC (pg mL−1) with a correlation coefficient of 0.997, respectively. Some previous immunosensors29,30 and voltammetric analysis31 also exhibited two slopes of calibration curves. For this work, the possible reason for the presence of two linear ranges was that the accumulation of the conductive Ag@BSA microspheres with the high concentration of HIgG enhanced the charge transfer in electrochemical oxidation of nanosilver, and therefore improved the sensitivity of the immunoassay. The detection limit was estimated to be 0.5 fg mL−1 at a signal-to-noise of 3. Based on the comparison listed in Table 1, we can find that this immunosensor exhibited an ultralow detection limit which is one order of magnitude lower than the previous immunosensor using ferrocene tagged peptide nanowire as probes9 or thionine doped mesoporous ZnO,32 and was much lower than that of the immunosensor based on in situ deposited polyaniline33 or gold nanoparticles,34 as well as the newly-reported label-free electrochemical immunosensor.4,35
lgC (pg mL−1) with a correlation coefficient of 0.994 and I = 18.01 + 46.97lgC (pg mL−1) with a correlation coefficient of 0.997, respectively. Some previous immunosensors29,30 and voltammetric analysis31 also exhibited two slopes of calibration curves. For this work, the possible reason for the presence of two linear ranges was that the accumulation of the conductive Ag@BSA microspheres with the high concentration of HIgG enhanced the charge transfer in electrochemical oxidation of nanosilver, and therefore improved the sensitivity of the immunoassay. The detection limit was estimated to be 0.5 fg mL−1 at a signal-to-noise of 3. Based on the comparison listed in Table 1, we can find that this immunosensor exhibited an ultralow detection limit which is one order of magnitude lower than the previous immunosensor using ferrocene tagged peptide nanowire as probes9 or thionine doped mesoporous ZnO,32 and was much lower than that of the immunosensor based on in situ deposited polyaniline33 or gold nanoparticles,34 as well as the newly-reported label-free electrochemical immunosensor.4,35
 | ||
| Fig. 4 ASVs of the immunosensing system incubated in HIgG solution with different concentrations (A) and calibration curves for HIgG determination (B) (n = 3 for error bars). | ||
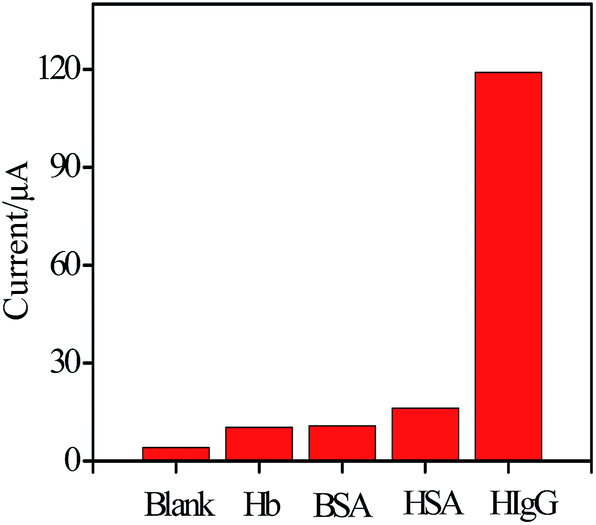 | ||
| Fig. 5 ASV response of the immunosensor to blank control, 10 pg mL−1 Hb, 10 pg mL−1 HSA, 1% BSA and 0.1 ng mL−1 HIgG. | ||
| Sensing platforma | Linear range (pg mL−1) | Detection limit (pg mL−1) | Ref. |
|---|---|---|---|
| a SPCE: screen-printed carbon electrode; GN: graphene; PNW-Fc: ferrocene-peptide nanowire; AgNW: silver nanowire; HRP: horseradish peroxidase; TH: thionine; Chit: chitosan; PDA: polydopamine; CNTs: carbon nanotubes; CuSNWs: CuS nanowires; AA: ascorbic acid. | |||
| GCE/AuNPs-GN-Ab1/HIgG/Ab2-PNW-Fc | 0.01–100 | 0.005 | 9 |
| GCE/AgNW-Chit/Ab1/HIgG/HRP-Ab2-ZnO-TH | 10–200000 |
4 | 32 |
| SPCE/GN/AuNPs/Ab1/HIgG/Au NP-Ab2-HRP | 20–500000 |
9.7 | 33 |
| SPCE/CNT-Ab1/HIgG/Ab2-AuNPs-PDA/silica | 10–10000 |
6.9 | 34 |
| GCE/GN-CNTs-Pd/Ab1/HIgG | 10–25000 |
3.3 | 4 |
| GCE/CuSNWs-Chit/Ab1/HIgG (AA in solution) | 1–320000 |
0.1 | 35 |
| GCE/β-CD-AuNPs/Ab1/HIgG/Ab2-BSA@Ag | 0.001–10 | 0.0005 | This work |
To evaluate specificity of the developed immunosensor for the detection of HIgG, the immunosensor was incubated with several other low-abundance proteins in serum such as hemoglobin (10 pg mL−1), human serum albumin (10 pg mL−1), BSA (1%). As depicted from Fig. 5, the current response for the target analyte was much higher than those of others, indicating that the immunosensor possesses a good selectivity in detecting HIgG.
The stability of Ag@BSA and Ag@BSA-antibodies are both good. Ag@BSA particles dispersed in aqueous solution remained their initial shape and particles size after twelve months storage in 4 °C. Ag@BSA-antibodies can be applied for the immunosensing within three months of storage in 4 °C. And the ASV signal of the immunosensor for 0.1 ng mL−1 of HIgG retained 96% of its initial current after the electrode was stored at 4 °C for four weeks. The good stability of the immunosensor was contributed to the good biocompatibility and effective antibody immobilization of Ag@BSA.
Analysis of real samples
The determination of HIgG in human serum sample was employed by standard addition method to demonstrate the practical application of the proposed immunosensor. The results were summarized in Table 2. It showed that the recovery ranged from 91.3% to 109.0% and the RSD was from 3.3% to 5.9%. These results indicated acceptable reliability of the method for practical applications.| Initial HIgG in sample | Measured after addition | ||||
|---|---|---|---|---|---|
| C (pg mL−1) | RSD (%, n = 5) | Added (pg mL−1) | Found (pg mL−1) | Recovery (%) | RSD (n = 5) |
| 410.00 | 2.6 | 0.1 | 410.11 | 109.0 | 5.9 |
| 10 | 419.13 | 91.3 | 3.3 | ||
| 500 | 892.73 | 96.5 | 4.6 | ||
Conclusions
In this work, a novel sandwich-type immunosensor for sensitive electrochemical determination of HIgG was constructed. Ag@BSA microspheres worked as the antibody carriers and electroactive probes. The immunosensor exhibited an ultralow detection limit, wide dynamic range and satisfied recovery in human serum. Therefore, this method provides a promising strategy for protein biomarker determination in clinical applications.Acknowledgements
The authors gratefully acknowledge the financial support of the National Science Foundation of China (No. 21275116).References
- M. A. Slatter and A. R. Gennery, Clin. Exp. Immunol., 2008, 152, 389–396 CrossRef CAS PubMed
.
- A. R. Gonzalez-Quintela, A. F. Gude, J. Campos, J. Rey and L. M. Meijide, et al., Clin. Exp. Immunol., 2008, 151, 42–50 CrossRef CAS PubMed
- R. Li, K. B. Wu, C. X. Liu, Y. Huang, Y. Y. Wang and H. G. Fang, et al., Anal. Chem., 2014, 86, 5300–5307 CrossRef CAS PubMed
- L. Liu, Y. Li, L. Tian, T. Guo, W. Cao and Q. Wei, Sens. Actuators, B, 2015, 211, 170–176 CrossRef CAS
- G. S. Lai, L. L. Wang, J. Wu, H. X. Ju and F. Yan, Anal. Chim. Acta, 2012, 721, 1–6 CrossRef CAS PubMed
- S. Weng, Q. Liu, C. Zhao, G. Hong, Z. Jiang and L. Lin, Sens. Actuators, B, 2015, 216, 307–315 CrossRef CAS
- B. V. Chikkaveeraiah, A. A. Bhirde, N. Y. Morgan, H. S. Eden and X. Chen, ACS Nano, 2012, 6, 6546–6561 CrossRef CAS PubMed
- L. Ma, D. Ning, H. Zhang and J. Zheng, Biosens. Bioelectron., 2015, 68, 175–180 CrossRef CAS PubMed
- Y. Y. Ding, D. Li, B. Li, K. Zhao, W. Du and J. Y. Zheng, et al., Biosens. Bioelectron., 2013, 48, 281–286 CrossRef CAS PubMed
- S. H. Jenkins, W. R. Heineman and H. B. Halsall, Anal. Biochem., 1988, 168, 292–299 CrossRef CAS PubMed
- G. Lai, H. Zhang, J. Yong and A. Yu, Biosens. Bioelectron., 2013, 478, 178–183 CrossRef PubMed
- J. Zhou, J. Tang, G. N. Chen and D. P. Tang, Biosens. Bioelectron., 2014, 54, 323–328 CrossRef CAS PubMed
- C. Y. Hu, D. P. Yang, Z. H. Wang, P. Huang, X. S. Wang, D. Chen, D. X. Cui, M. Yang and N. Q. Jia, Biosens. Bioelectron., 2013, 41, 656–662 CrossRef CAS
- S. B. He, H. H. Deng, A. L. Liu, G. W. Li, X. H. Lin, W. Chen and X. H. Xia, ChemCatChem, 2014, 6, 1543–1548 CrossRef CAS
- C. Y. Hu, D. P. Yang, F. J. Zhu, F. J. Jiang, S. Y. Shen and J. L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4170–4178 CAS
- C. Y. Hu, D. P. Yang, Z. Y. Wang, L. L. Yu, J. L. Zhang and N. Q. Jia, Anal. Chem., 2013, 855, 200–5206 Search PubMed
- C. Y. Hu, D. P. Yang, K. Xu, H. M. Cao, B. N. Wu, D. X. Cui and N. Q. Jia, Anal. Chem., 2012, 84, 10324–10331 CrossRef CAS PubMed
- H. M. Cao, D. P. Yang, D. X. Ye, X. X. Zhang, X. E. Fang, S. Zhang, B. H. Liu and J. L. Kong, Biosens. Bioelectron., 2015, 68, 329–335 CrossRef CAS PubMed
- C. Ma, W. P. Li, Q. K. Kong, H. M. Yang, Z. Q. Bian, X. R. Song, J. H. Yu and M. Yan, Biosens. Bioelectron., 2015, 63, 7–13 CrossRef CAS PubMed
- J. Gao, Z. K. Guo, F. J. Su, L. Gao, X. H. Pang, W. Cao, B. Du and Q. Wei, Biosens. Bioelectron., 2015, 63, 465–471 CrossRef CAS PubMed
- Q. F. Li, D. P. Tang, F. M. Lou, X. M. Yang and G. N. Chen, ChemElectroChem, 2014, 1, 441–447 CrossRef CAS
- X. Wang, X. Li, C. Luo, M. Sun, L. Li and H. Duan, Electrochim. Acta, 2014, 130, 519–525 CrossRef CAS
- J. Wen, S. Zhou and Y. Yuan, Biosens. Bioelectron., 2014, 52, 44–49 CrossRef CAS PubMed
- T. Huang, F. Meng and L. M. Qi, J. Phys. Chem. C, 2009, 113, 13636–13642 CAS
- A. Gebregeorgis, C. Bhan, O. Wilson and D. Raghavan, J. Colloid Interface Sci., 2013, 389, 31–41 CrossRef CAS PubMed
- P. Huang, D. P. Yang, C. Zhang, J. Lin, M. He, L. Bao and D. Cui, Nanoscale, 2011, 3, 3623–3626 RSC
- G. Wang, X. Gang, X. Zhou, G. Zhang, H. Huang, X. Zhang and L. Wang, Talanta, 2013, 103, 75–80 CrossRef CAS
- Y. Zhu, P. Chandra and Y. B. Shim, Anal. Chem., 2013, 85, 1058–1064 CrossRef CAS PubMed
- D. Feng, L. Li, X. Fang, X. Han and Y. Zhang, Electrochim. Acta, 2014, 127, 334–341 CrossRef CAS
- X. Gao, Y. Zhang, Q. Wu, H. Chen, Z. Chen and X. Lin, Talanta, 2011, 85, 1980–1985 CrossRef CAS PubMed
- Y. Fan, J. H. Liu, C. P. Yang, M. Yu and P. Liu, Sens. Actuators, B, 2011, 157, 669–674 CrossRef CAS
- X. Cao, S. Liu, Q. Feng and N. Wang, Biosens. Bioelectron., 2013, 49, 256–262 CrossRef CAS PubMed
- G. Lai, H. Zhang, T. Tamanna and A. Yu, Anal. Chem., 2014, 86, 1789–1793 CrossRef CAS
- G. Lai, H. Zhang, J. Yong and A. Yu, Biosens. Bioelectron., 2013, 47, 178–183 CrossRef CAS PubMed
- N. Wang, C. Gao, Y. Han, X. Huang, Y. Xu and X. Cao, J. Mater. Chem. B, 2015, 3, 3254–3259 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22654f |
| This journal is © The Royal Society of Chemistry 2015 |