
Self-repairing nonfouling polyurethane coatings via 3D-grafting of PEG-b-PHEMA-b-PMPC copolymer
Abstract
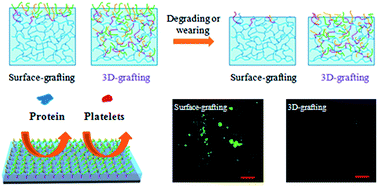
Durability of nonfouling coatings is a critical problem to be solved for their practical application. In this paper, self-repairing nonfouling polyurethane (PU) coating was fabricated by spraying of a triblock copolymer of polyethylene glycol (PEG), 2-hydroxyethyl methacrylate (HEMA) and 2-methacryloyloxyethyl phosphorylcholine (MPC) on pre-cured acrylic-based PU coatings. The triblock copolymer was synthesized through living radical polymerization mediated by an atom transfer radical polymerization. Anchoring of the copolymer took place both on the surface and in the interior (namely, 3D-grafting) of PU coatings through the diffusion of the copolymer and subsequently the chemical reaction between the pendent hydroxyl group of the copolymer and the remaining isocyanate groups in the pre-cured PU coatings. This 3D grafting procedure is rather simple and favorable for large-area application. The 3D-grafted PU coating has higher hydrophilicity (water contact angle: 33°). In contrast, if the triblock copolymer was used as an additive in PU coatings, the hydrophilicity of the PU coating did not obviously change. The 3D-grafted PU coating effectively inhibited the adhesion of protein and human platelet cells due to the synergistic effect of PEG and MPC. Moreover, after the coatings were detached and mechanically damaged, the surfaces can restore their hydrophilicity and possess better long-term anti-fouling ability than the control surface-grafted coatings underwater.
 Please wait while we load your content...
Please wait while we load your content...

