
Enhanced mechanical properties of epoxy nanocomposites based on graphite oxide with amine-rich surface
Ya Zhou,
Le Li,
Yang Chen,
Huawei Zou* and
Mei Liang*
The State Key Lab of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: hwzou@163.com; liangmeiww@163.com; Fax: +86-28-85402465; Tel: +86-28-85408288
First published on 12th November 2015
Abstract
Functional graphite oxide (DGO) with amine-rich surface was synthesized through chemically grafting flexible poly(oxypropylene)diamine, and its epoxy (EP) composites were prepared. Fourier transform infrared spectra (FTIR) etc. confirmed the realization of chemical functionalization of DGO. The covalent functionalization of graphite oxide (GO) with poly(oxypropylene)diamine was favorable to its homogeneous dispersion in epoxy matrix. Meanwhile, the strong covalent interface formed between epoxy and DGO promoted the stress transfer. The addition of 0.3 wt% DGO increased the tensile strength, flexural strength, elongation at break and toughness of the epoxy resins by 20%, 40%, 90% and 145%, respectively. This showed higher improvements than those addition of GO. Therefore, significant improvements both in the strength and toughness of epoxy nanocomposites were achieved by the addition of trace DGO.
1. Introduction
Epoxy resin is a matrix with excellent physical and chemical properties after curing, such as low cure shrinkage, compatibility with a great number of materials, high mechanical strength, adhesion, chemical resistance and good electrical insulation, giving rise to a wide use in composite materials.1 However, epoxy resins are brittle and have poor resistance to the crack propagation owning to its high cross-link density as thermosetting materials.2,3 Various reinforcements used fail to achieve the improvement of toughness and strength of polymer at the same time, since the two properties are almost mutually exclusive.4–7 It has recently been reported that the incorporation of graphite and graphite oxide could enhance the toughness of thermosetting materials successfully.8–10Graphene, consisting of a single-layered sheet of hexagonally arrayed sp2 hybridized carbon atoms, has a combination of exceptional mechanical, electrical and thermal properties, such as a Young's modulus of 1 TPa and breaking strength of 130 GPa.11–15 Owing to its excellent properties, polymer nanocomposites with graphite and its derivatives have attracted tremendous attention, both in industry and in academia.16,17 Meanwhile, to take full advantage of the potential of graphite as nanofiller, two critical factors should be taken into consideration. One is the homogeneous dispersion of graphite into the matrix, and the other is strong interfacial interactions required between the graphite and the matrix.18 Excellent dispersion quality ensures a high specific surface area which can guarantee the strong filler/matrix adhesion in the composites.19 Meanwhile, the strong interfacial interaction is beneficial to efficient stress transfer.10 However, due to the strong van der Waals forces, pristine graphite has a high tendency to agglomerate, which restricts its dispersion in polymer matrix seriously.20 In order to deal with the problems above, the oxidation of graphite that leads to the formation of graphite oxide (GO) is proposed. The functional groups (epoxide, hydroxyl and carboxyl groups) of GO can change the van der Waals significantly and afford active sites for further functionalization. This is beneficial to improve the solubility and processability as well as enhance the interactions with organic polymers.21,22 By far, various functionalization methods have been reported, including covalent and non-covalent functionalization.23
Considerable work has been carried out on the enhancement of the mechanical and thermal properties of GO-based epoxy (EP) composites.24–28 However, few studies have reported that the epoxy nanocomposites could achieve the improvement of strength and toughness simultaneously with the existence of functional graphite oxide (DGO). Zaman et al. achieved the surface modification of graphene with 4,4-methylene diphenyl diisocyanate (MDI), which led to improved dispersion and interface interaction in the epoxy. The filler/epoxy nanocomposites showed a further 96.1% increase of fracture energy release rate over the unmodified nanocomposite, but this was accompanied by a reduction of tensile strength.29 Chatterjee et al. have reported a maximum increase of 8% both in modulus and hardness for epoxy composite with 1.5 wt% of GO functionalized with dodecylamine (DDA). The fracture toughness of the filler/epoxy increased by 66% with the addition of 0.1 wt% functionalized-GO, but no more increase was observed as the loading of filler increased.30 Several studies reported that the presence of the amine groups on carbon materials could improve the adhesion of the filler to epoxy matrix, since these functional groups had good compatibility with this polymer system.31–33 Based on these ideas, the curing agent poly(oxypropylene)diamine D2000 is selected as the grafting molecule. On the one hand, it is a kind of flexible molecule and proposed to be compatible well with epoxy. On the other hand, the unreacted amino groups of poly(oxypropylene)diamine may form covalent bonding with epoxy resin, improving the interfacial interaction between graphene and polymer matrix. Meanwhile, the surface functionalization process is relatively simple and easy to operate. In previous studies, we investigated the effects brought by incorporating GO and DGO sheets in the curing process of EP, which established a basis for optimizing the performance of graphene/epoxy nanocomposites.34
In this work, the functional GO with amine-rich surface was prepared by introducing flexible poly(oxypropylene)diamine onto the GO surface, looking forward to improving the dispersion and reinforcing the interaction between the graphene and the matrix simultaneously. Compared with neat epoxy system and GO/epoxy system, the aim of our work was to study the incorporation of amine-rich GO surface on the influence of properties of epoxy nanocomposites. Besides, this study provided a better understanding of the relationship between structure and properties of the composites, which enabled improvement of the composites functionalities.
2. Experimental section
2.1 Materials
The diglycidyl ether of bisphenol A type (E51) epoxy resin was provided by Deyuan Chemistry Plant, China, with an epoxide equivalent weight of 210–240 g per equiv. The density is 1.18 g cm−3 at 25 °C and the viscosity is 2500 mPa s at 40 °C. The curing agent was 4,4-diamino diphenyl methane (DDM), which was provided in analytical grade by Shanghai crystal neat reagent Co. (Aladdin Reagent Co., China). The natural graphite was provided by Qing-dao Dongkai Graphite Co. Ltd. (Qingdao, China). The graphite has a particle size of 45 μm with a density of 2.25 g cm−3. Poly(oxypropylene)diamine were obtained from Huntsman Corporation (The Woodlands, Texas, USA), with average weight molecular weight of 2000 g mol−1 (D2000). Sodium nitrate (NaNO3), potassium permanganate (KMnO4), 30 wt% hydrogen peroxide (H2O2) solution, 98% H2SO4 and 37 wt% HCl were supplied by Shanghai SSS Reagent Company.2.2 Preparation of graphite oxide (GO)
Graphite oxide was prepared from purified natural graphite by Hummers method.35,36 About 5 g graphite powder and 2.5 g NaNO3 were added into 115 mL cooled concentrated H2SO4. About 15 g KMnO4 was gradually dropped into the above liquid with stirring and the mixture was kept below 20 °C during this process. Then the mixture was heated to 35 °C and stirred for 30 min. After which, 230 mL deionized water (DI-water) was slowly added, increasing the temperature to 98 °C and maintaining it for about 15 min. The reaction was terminated by adding 140 mL distilled water and 10 mL 30% H2O2 solution. The resulting mixture was filtrated and washed with diluted HCl solution for several times until no sulfate ion could be detected by BaCl2. Finally, it was dried in vacuum oven at 60 °C for overnight.2.3 Synthesis of functional graphite oxide (DGO) grafted by poly(oxypropylene)diamine
GO (0.2 g) was dispersed in 200 mL of DMF (dimethyl formamide) and exfoliated by ultrasonication in a round-bottom flask. Then, poly(oxypropylene)diamine D2000 and DMAP (4-dimethylaminopyridine) (with the ratio of GO, D-2000 and DMAP of 0.2 g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 mmol:6 mmol) were mixed in a flask and stirred at 90 °C for 24 h. After cooling to room temperature, excess D2000 was removed by washing with a mixture of DCM (dichloromethane) and petroleum ether several times. The remaining solid was separated by filtration using a 0.2 μm membrane filter. The collected solid was again washed with ethanol several times and dried at 60 °C under vacuum to generate DGO powders. The process to prepare DGO is shown in the Scheme 1.
:6 mmol:6 mmol) were mixed in a flask and stirred at 90 °C for 24 h. After cooling to room temperature, excess D2000 was removed by washing with a mixture of DCM (dichloromethane) and petroleum ether several times. The remaining solid was separated by filtration using a 0.2 μm membrane filter. The collected solid was again washed with ethanol several times and dried at 60 °C under vacuum to generate DGO powders. The process to prepare DGO is shown in the Scheme 1.
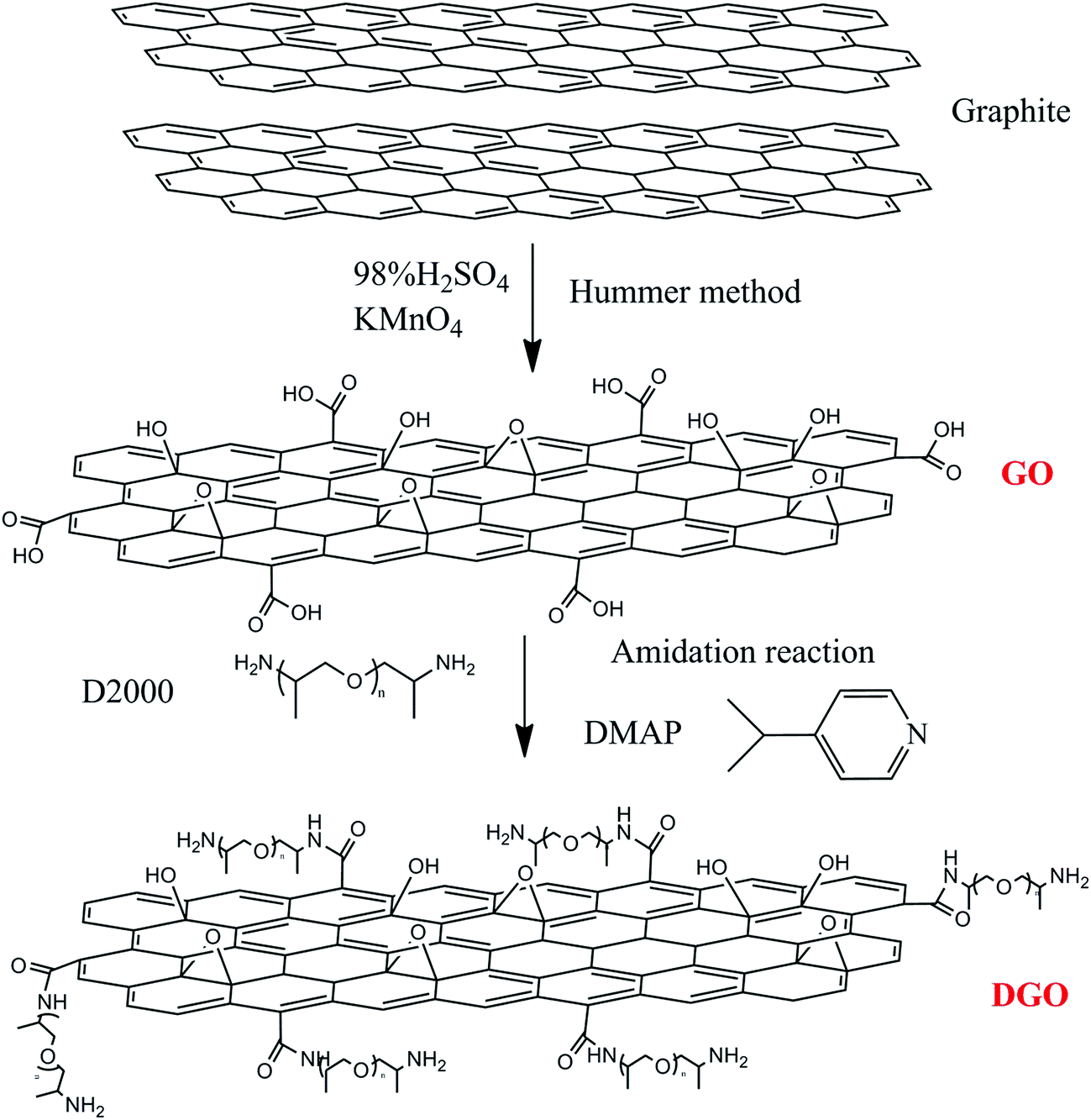 | ||
| Scheme 1 Reaction scheme of grafting poly(oxypropylene)diamine onto GO surface. | ||
2.4 Nanocomposites preparation
Graphite oxide (GO) was incorporated into the epoxy resin in certain proportion. The detailed preparation process of GO/EP (GO and epoxy nanocomposites) was as follows. The particles were first dispersed in acetone with the assistance of sonication at room temperature for 1 h. The solutions were then mixed with the epoxy by sonication for 30 min to ensure dispersion homogeneity. The solvent was removed in a vacuum oven at 80 °C for 5 h. Afterwards, DDM was added to the mixture with a high speed mechanical stirrer at 2500 rpm to obtain a homogeneous mixture. Weight ratio of epoxy/hardener (DDM) was 4:1 (stoichiometric). The mixture was subsequently degassed under vacuum for about 20 min. Then the mixture was poured into a mold for curing at 100 °C for 1 h, 150 °C for 4 h and 180 °C for 1 h. For comparison, the samples designated as EP (neat epoxy) and DGO/EP (DGO and epoxy nanocomposites) were also prepared in the same procedure described for GO/EP.
2.5 Characterization methods
3. Result and discussion
3.1 Characterization of graphite oxide (GO) and functional graphite oxide (DGO)
FTIR spectrum of GO in Fig. 1 shows the typical bands belonging to GO. The incorporating epoxide (C–O–C 1215 cm−1, 1055 cm−1), hydroxyl (3432 cm−1) and carboxyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1725 cm−1, C–OH 1645 cm−1) demonstrate the process of oxidation is successful. After grafting reaction, the FTIR spectrum of DGO appears some new and more intense peaks. The new bands observed for DGO at 2972, 2867, 1628, 1453, 1100 cm−1 can be attributed to the vibrations of C–H (CH3), C–H (CH2), CO–NH, C–H (CH2), C–O, respectively. These significant existence of new bands indicates the possibility of the grafted poly(oxypropylene)diamine chains on GO (note that this sample was exhaustively washed to remove the free poly(oxypropylene)diamine chains adsorbed on GO sheets prior to the FTIR tests). Especially, the existence of the characteristic peak at 1628 cm−1 directly identifies the formation of CO–NH bond between the poly(oxypropylene)diamine and the carboxylic groups on GO.
O 1725 cm−1, C–OH 1645 cm−1) demonstrate the process of oxidation is successful. After grafting reaction, the FTIR spectrum of DGO appears some new and more intense peaks. The new bands observed for DGO at 2972, 2867, 1628, 1453, 1100 cm−1 can be attributed to the vibrations of C–H (CH3), C–H (CH2), CO–NH, C–H (CH2), C–O, respectively. These significant existence of new bands indicates the possibility of the grafted poly(oxypropylene)diamine chains on GO (note that this sample was exhaustively washed to remove the free poly(oxypropylene)diamine chains adsorbed on GO sheets prior to the FTIR tests). Especially, the existence of the characteristic peak at 1628 cm−1 directly identifies the formation of CO–NH bond between the poly(oxypropylene)diamine and the carboxylic groups on GO.
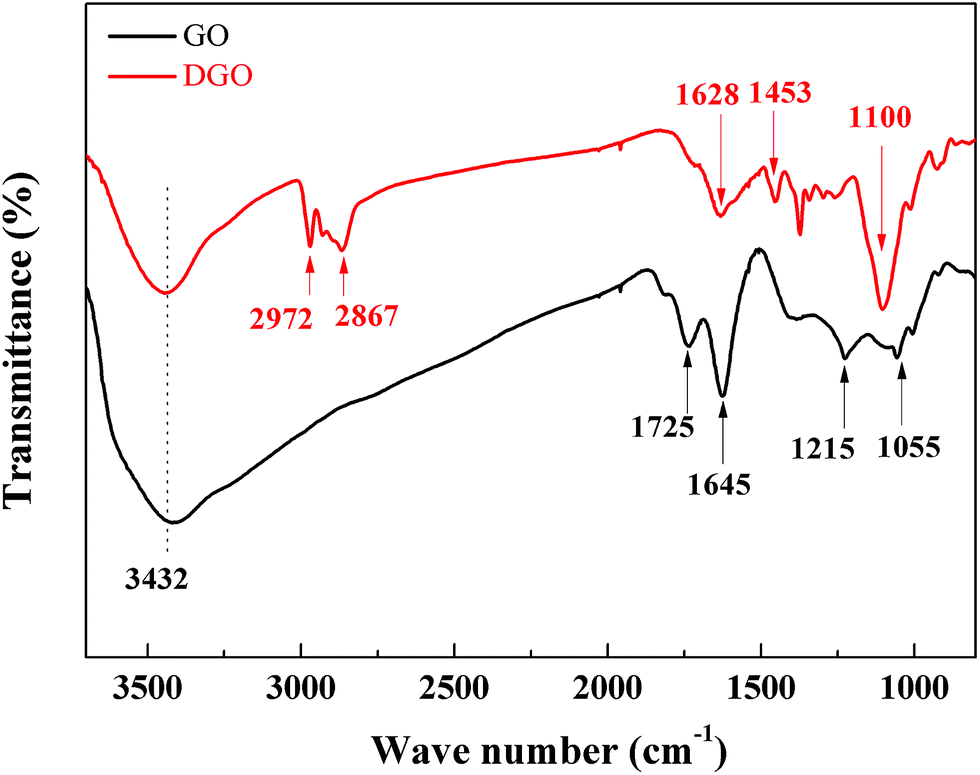 | ||
| Fig. 1 FTIR spectrum of GO and DGO. | ||
Fig. 2 shows the representative AFM images of GO and DGO sheets. The GO sheets show a smooth clean surface (Fig. 2a), and their length sizes range from 0.1 μm to 1.5 μm with a mean thickness of 0.936 nm, consistent with the reported results.37,38 After grafting poly(oxypropylene)diamine to GO, the mean thickness of DGO increases to 2.656 nm. An additional 1.72 nm increase primarily originates from the contribution of poly(oxypropylene)diamine, which is similar to the result reported by Lomeda et al.38 Besides that, the surface of the DGO is rougher than GO, which is also attributed to the bonding of poly(oxypropylene)diamine. The change of nano-layer morphology and thickness indicates the successful functionalization and the uniform distribution of grafting molecules on the GO surface.
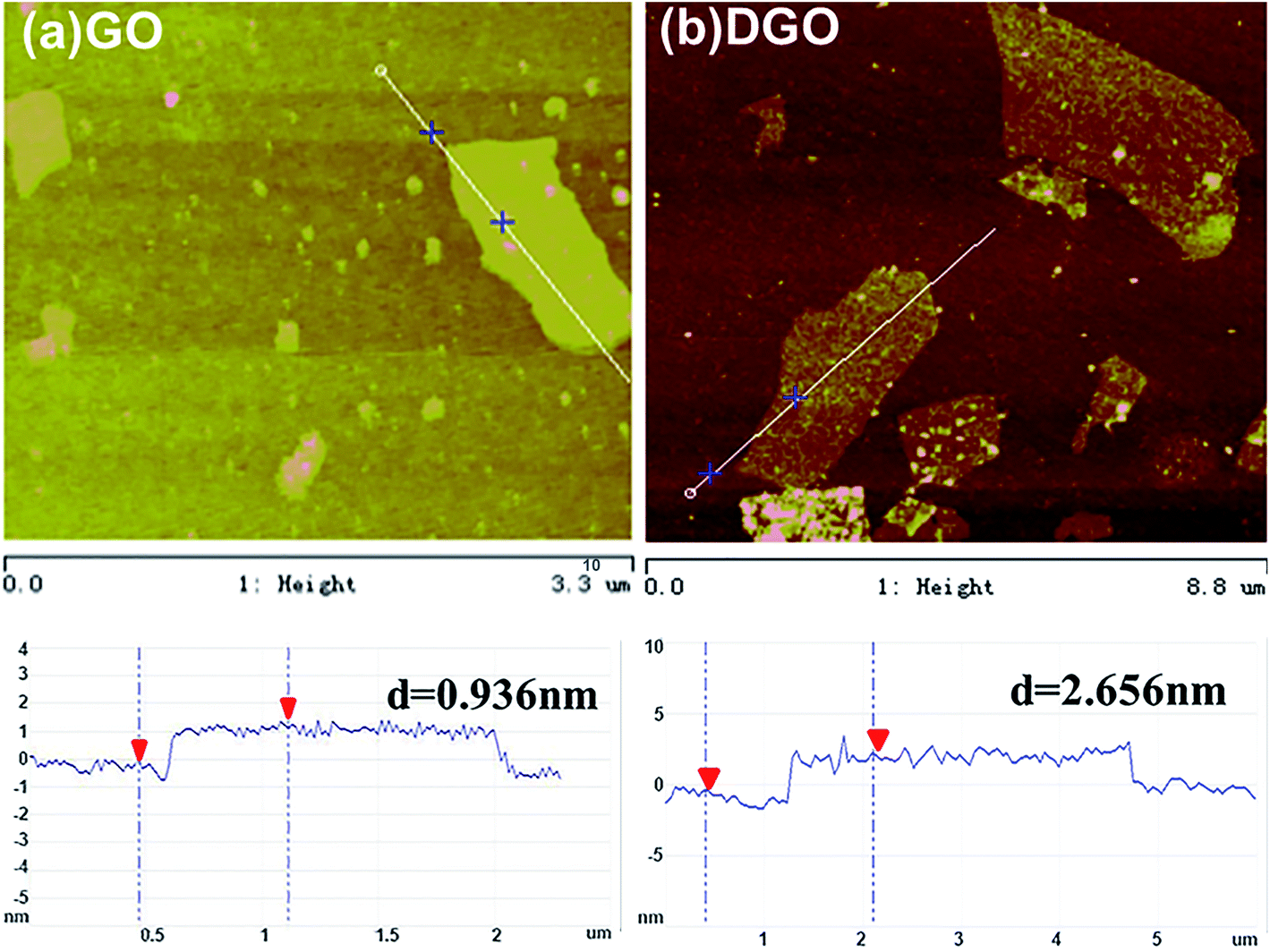 | ||
| Fig. 2 Tapping mode AFM images of (a) GO and (b) DGO. | ||
Raman spectra of GO and DGO are shown in Fig. 3, as it is a convenient and powerful tool to probe the structure of GO and DGO. Both oxidation and grafting reaction generally introduce defects in graphite sheets.39 The defects are often pronounced in spectroscopic data, from which additional structural information of samples can be derived. The D band originates from defects inherent in the graphite and the edge effect of graphite crystallites, while the G band arises primarily from the presence of a sp2 carbon network. Compared with GO, the D peak of DGO red-shifts from 1340 cm−1 to 1328 cm−1, which arises primarily from the symmetry breaking at the edges due to poly(oxypropylene)diamine grafting.40 It is also worth noting that the G band shifts from 1584 cm−1 to 1595 cm−1, indicating a larger interlayer space for DGO.41 The 2D band, relative to stacking order in pristine graphite, generates a peak at about 2700 cm−1.42 However, it is barely visible for both GO and DGO, implying the decrease of stacking order. Meanwhile, the intensity ratio of D and G bands (ID/IG), related to the disordered or ordered crystal structure, can be seen as a measure of the extent of disorder of carbon.43 A simple method to assess the density of graphite defects is to calculate the crystallite size La. As proposed,  (λlaser is the laser excitation wave length with the value of 633 nm).42 In our case, the ID/IG ratio of GO and DGO is 1.03 and 1.20 respectively. Hence, La of GO and DGO is 39.69 and 32.11 nm, respectively. The La of DGO reduces from 39.69 to 32.11 nm by grafting, indicating the defect density is significant and further supporting the assertion that covalent bond was formed between GO and poly(oxypropylene)diamine.
(λlaser is the laser excitation wave length with the value of 633 nm).42 In our case, the ID/IG ratio of GO and DGO is 1.03 and 1.20 respectively. Hence, La of GO and DGO is 39.69 and 32.11 nm, respectively. The La of DGO reduces from 39.69 to 32.11 nm by grafting, indicating the defect density is significant and further supporting the assertion that covalent bond was formed between GO and poly(oxypropylene)diamine.
 | ||
| Fig. 3 Raman spectra of GO and DGO nanosheets. | ||
Further proof of the chemical transformations of poly(oxypropylene)diamine induced by grafting was obtained by thermogravimetric analysis (TGA). The plots for the GO and DGO in N2 atmosphere were investigated and the results are shown in Fig. 4a and b. For GO (Fig. 4a), the mass loss starts below 100 °C, which is ascribed to the evaporation of adsorbed water.44–46 As it is known, GO sheets bear oxygen-containing functional groups with strong hydrophilic properties, so that water molecules could be bound tightly into their stacked structure.47 The main mass loss occurring at around 200 °C is presumably due to pyrolysis of the labile oxygen-containing functional groups to yield CO, CO2 and steam.44,48 However, the steady loss observed for temperatures above 250 °C and up to 800 °C is assigned to the elimination of more stable oxygen functionalities.44,46
 | ||
| Fig. 4 TGA (a) and DTG (b) curves of GO and DGO in N2 atmosphere. | ||
In the case of DGO (Fig. 4b), the peak representing maximum weight loss rate shifts to 373 °C, compared with that of GO (206 °C). It implies that the labile oxygen functional groups of GO have partly been replaced by poly(oxypropylene)diamine to increase the thermostability of GO. That is to say, the mass loss between 300 °C to 400 °C is presumably assigned to the decomposition of the grafting poly(oxypropylene)diamine on DGO.47 As shown in Fig. 4a, the residual char of GO is 46% at 800 °C, which is 29.8% higher than that of DGO. This indicates that DGO contains less carbon skeleton than GO due to the grafting of poly(oxypropylene)diamine, on the condition of the same weight.
3.2 Characterization of epoxy nanocomposites
SEM characterization of freeze-fractured specimens has been done to understand the dispersion state of pristine and functional GO in epoxy matrix, and the results are displayed in the below. In Fig. 5a, the fracture surface of the neat resin is quite smooth and mirror-like. In contrast, the untreated GO are dispersed in the epoxy matrix mainly with the form of big and irregular protuberance (Fig. 5b). However, protuberances are in few and small nanometer ranges in case of modified GO filled in EP (DGO/EP) (Fig. 5c), confirming much better dispersion of DGO in epoxy matrix. The improved compatibility and dispersion of functional GO in the epoxy matrix (DGO/EP) is mainly attributed to the grafting molecules, which leads to a strong interfacial interaction between the amine groups on the GO and epoxy matrix. | ||
| Fig. 5 SEM images of the freeze-fractured surface of the neat epoxy (a) and nanocomposites: (b) 0.3 wt% GO/EP, and (c) 0.3 wt% DGO/EP. | ||
The fractured surfaces after tensile tests are showed to explore the microstructure of the nanocomposite samples and the interface between the sheets and the epoxy matrix. As shown in Fig. 6a, the fracture surface of the pure epoxy is quite smooth, revealing the brittleness and its nature of weak resistance to crack initiation and propagation. On the contrary, the composites with GO and DGO exhibit rough surfaces, as shown in Fig. 6b and c. For the GO/epoxy nanocomposites, the SEM image presents typical aggregates of sheets in the middle of the surface, which is consistent with the freeze-fracture observations. Some gaps between GO sheets and matrix can also be found on the surface (see the red circle in the Fig. 6b), indicating the relatively weak interfacial bonding between the GO and the epoxy matrix. Such aggregates of GO sheets and poor sheet/matrix interface can cause a stress concentration during the fracture process, impairing stress transfer from the matrix to the sheets. Comparatively, no obvious aggregates and clear gaps between the DGO sheets and the matrix are observed on the fractured surface (Fig. 6c), revealing that the DGO sheets are well dispersed and embedded in epoxy. The improved interfacial interaction between sheets and matrix is primarily attributed to the functionalization of poly(oxypropylene)diamine. Meanwhile, the observed tortuous structures with hackles and ribbons reflect an increase of the energy dissipation during the fracture process and a higher fracture toughness value as shown in Fig. 7.
 | ||
| Fig. 6 SEM fractographs of the neat epoxy (a) and nanocomposites: (b) 0.3 wt% GO/EP and (c) 0.3 wt% DGO/EP after tensile tests (the gaps marked in red circle indicate the delamination of GO sheets due to the weak interface). | ||
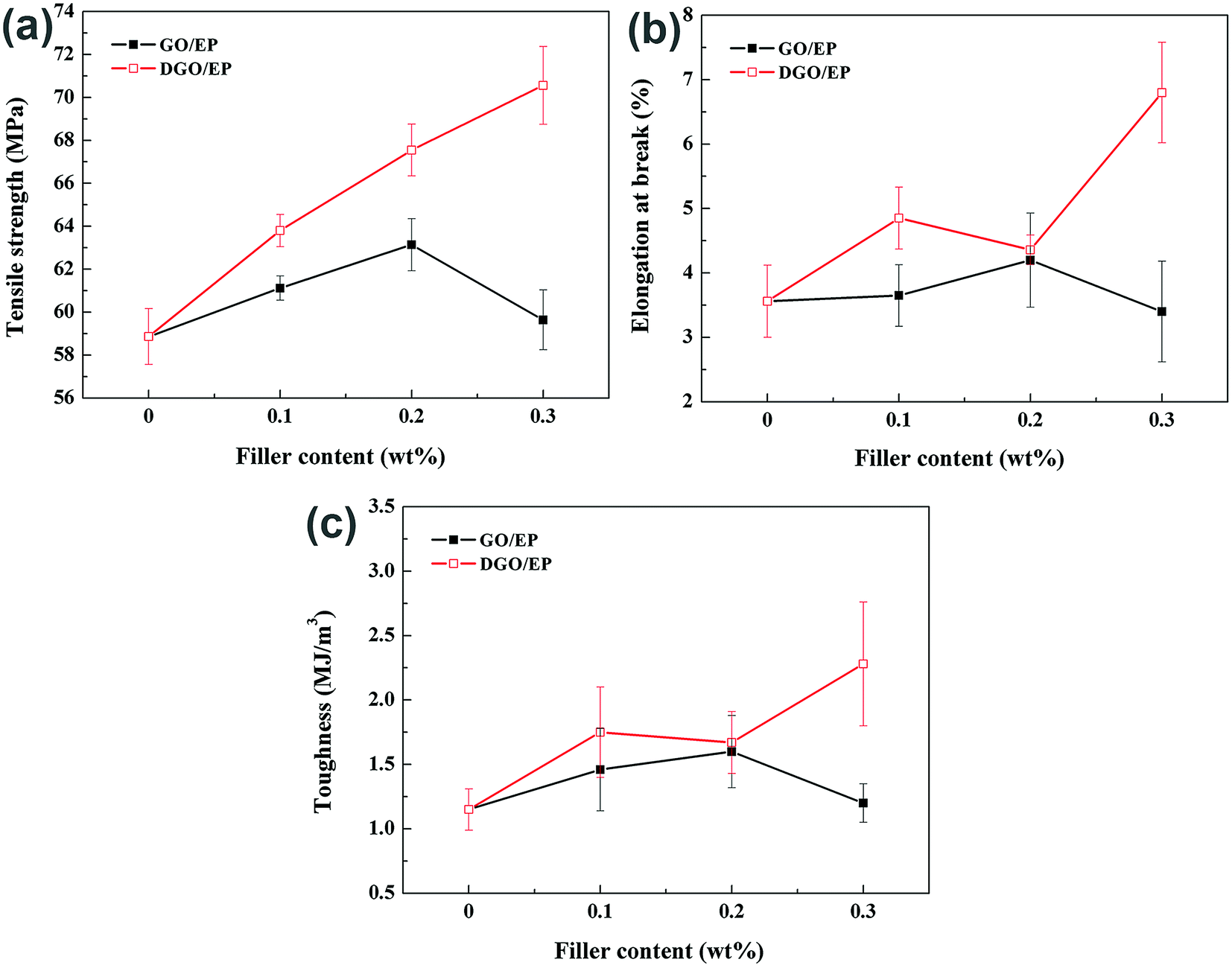 | ||
| Fig. 7 Tensile properties of neat epoxy and epoxy nanocomposites. | ||
The full dataset for neat epoxy and its nanocomposites, including tensile strength, elongation at break and toughness (calculated by the area under the stress–strain curve) are listed in Fig. 7. The tensile strength of GO/EP containing low quantities of GO (0.2 wt%) shows only an 7.3% increase over the tensile strength of neat EP. Further increasing the GO content from 0.2 to 0.3 wt% decreases the tensile strength from 7.3% to 1.3% over that seen for the neat EP. A similar trend was also found for the elongation at break and toughness of the GO/EP composite material. This occurs because further loading causes the GO sheets to stack together, reducing the improvement of mechanical properties. Consequently, we attribute the limited and non-continuous improvements in tensile properties of the GO/EP to two reasons, (i) the properties of GO deteriorate as GO sheets produce aggregation, because these clusters behave as micrometre-size fillers with relatively low surface area (ii) from SEM analysis, some GO sheets are easily pulled out, generating gaps in the EP matrix. This suggests the weak adhesion and poor compatibility between the GO sheets and the matrix. The weak interfacial interaction leads to low load transfer efficiency at the interface.
In comparison, the DGO/EP exhibit superior improvements in mechanical properties. The tensile strength of DGO/EP containing low quantities of DGO (0.2 wt%) showed 14.7% increase over that of neat EP. Different from the addition of GO, increasing the DGO content from 0.2 to 0.3 wt% further increases the tensile strength and elongation at break to 70.56 MPa and 6.80%, respectively. Compared to neat epoxy, the DGO/EP with only 0.3 wt% DGO exhibits a dramatic increase in tensile strength (approximately 20%) and a pronounced improvement in elongation at break (approximately 90%) as well, revealing that DGO has an exceptional capacity in strengthening and toughening epoxy. A 145% increase in toughness with the addition of 0.3 wt% DGO further indicates the toughening effect of DGO is significant. The notable and continuous reinforcement and toughening effects with the increasing addition of DGO drive us to explore the underlying reasons and mechanisms.
On one hand, the excellent mechanical properties of nanocomposites are attributed to the graphite itself with the ultrahigh surface area and superior mechanical properties as filler. The possible strengthening mechanism of graphite sheets is attributed to the better adhesion, which is caused by the enhanced mechanical interlocking between the wrinkled surface of graphite sheets and the epoxy chains.49 On the other hand, the poly(oxypropylene)diamine chain grafted at the GO surface can prevent stacking and aggregation of GO sheets, thus improving the dispersion state of the graphite sheets in EP matrix. And the amine functional groups of DGO afford an amine-rich environment near the sheets for epoxide groups of epoxy to form covalent bonding between the sheets and the matrix during the curing process. Consequently, the interfacial interaction between different components becomes stronger. Meanwhile, the formation of strong flexible interface due to long soft grafting chains plays a significant role in transferring load from epoxy matrix to graphite sheets.
Fig. 8 shows the results of three-point-bending tests of the neat epoxy and nanocomposite samples. As shown in Fig. 8a, the flexural strength of both DGO/EP and GO/EP increases with increasing the sheets content. However, DGO/epoxy nanocomposites show a more prominent increase both in strength and modulus at 0.1–0.3 wt%. The addition of 0.3 wt% DGO generally maintains the increased flexural modulus of composites, while the GO/EP presents an obvious decrease in flexural modulus at 0.3 wt% (in Fig. 8b). Compared to neat epoxy, the DGO/epoxy with only 0.3 wt% DGO exhibits a dramatic increase in flexural strength (approximately 42%) and an improvement in flexural modulus (approximately 15%), implying that DGO has an excellent capacity in strengthening epoxy.
 | ||
| Fig. 8 Flexural properties of neat epoxy and epoxy nanocomposites. | ||
The highly enhanced mechanical properties achieved in the DGO/epoxy composites can be attributed to (i) good compatibility and dispersion of DGO in the epoxy matrix (ii) strong interaction between the sheets and the matrix, originating from the wrinkled surface and poly(oxypropylene)diamine functionalization.
Fig. 9 and Table 1 show the dynamic mechanical properties of neat epoxy and its nanocomposites. It is recognized that the incorporation of GO and DGO more or less increase the storage modulus and Tg with various loading, except for the 0.3 wt% of GO. However, nanocomposites containing DGO exhibit a better enhancement for the same loading. As shown in Fig. 9a, the addition of 0.3 wt% DGO can maintain the increased storage modulus of composites, while the addition of 0.3 wt% GO fails to improve the storage modulus of epoxy. In contrast, the higher loading of GO deteriorates the rigidity of epoxy. This can be explained by the reinforcing effect of DGO and the improved interface quality after surface functionalization.
 | ||
| Fig. 9 The dynamic mechanical properties of neat epoxy and its nanocomposites. | ||
| wt% (GO/DGO) | Tg (°C) | |
|---|---|---|
| EP | 0 | 155 |
| GO/EP | 0.1 | 161 |
| 0.2 | 172.5 | |
| 0.3 | 149.5 | |
| DGO/EP | 0.1 | 163.5 |
| 0.2 | 174 | |
| 0.3 | 171 |
The glass transition temperature (Tg), reflecting the response of polymer segments to the imposed load significantly, is a macroscopic indication of the relaxation behavior of nanocomposites systems. And its magnitude is strongly influenced by embedded particles.25 As shown in Table 1, the Tg of DGO/EP containing only 0.2 wt% DGO (approximately 174 °C) was much higher than that of neat epoxy (155 °C), increasing by 19 °C. A similar trend could be found at 0.2 wt% loading of GO. The increased Tg value indicates that incorporating GO and DGO into the epoxy matrix both restrict the mobility of the polymer chains significantly so that the relaxation can only occur at higher temperature.
However, with higher content of GO introduced in epoxy, the Tg value of GO/EP demonstrated a significant decrease, even lower than that of neat epoxy. In contrast, the Tg of nanocomposites remained much higher value than that of neat epoxy (about an increase of 16 °C) at 0.3 wt% loading of DGO. Several reasons are probably ascribed to the above phenomena. First, in consistent with the results of SEM and tensile properties of nanocomposites, the aggregation of GO at higher loading leads to its poor dispersion in epoxy matrix and decreases the confinement effect, while the DGO remains a well-dispersed state in epoxy even with a higher loading. Moreover, introducing GO can disturb the curing reaction of epoxy, leading to low reaction conversion. This generally reduces the epoxy cross-linkage and increases the polymer segments mobility. On the other hand, embedded GO sheets may confine polymer segments and reduce the chain mobility. The balance of these two factors will influence the glass transition temperature. If the compromise factors tend to positive effect on polymer chain mobility, the Tg will decrease. The decreased Tg of GO/EP is consistent with Bao's results, who attributed this phenomenon to ‘‘cross-linking density reduction’’ effect.44 Besides, according to our previous study,34 we find that excess introduction of GO indeed interferences with epoxy curing reaction owing to the increment of viscosity, leading to a reduction in organic network density of epoxy. To sum up, an increase in the mobility of the epoxy segments results in the decreased Tg of GO/epoxy.
In the case of DGO/EP, the ends of the grafting chains participate in curing process of epoxy to form chemical bonds. It is the result of reaction between amine groups of poly(oxypropylene)diamine and epoxide groups of epoxy, which played the important role in remaining the cross-linking network density. The above reasons both explain the decreased Tg of GO/EP and the remained Tg of DGO/EP.
4. Conclusion
Functional graphite oxide (DGO) was successfully prepared through the amidation reaction between poly(oxypropylene)diamine and carboxylic acid groups on the surface of graphite oxide. After chemical modification, the functional graphite oxide sheets are compatible well in epoxy matrix, leading to a better dispersion and a stronger interface interaction between the filler and matrix. As to the nanocomposites, the enhanced tensile strength, flexural strength and toughness are shown with the addition of functional graphite oxide at the loading of 0.3 wt%, with 20%, 42% and 145% increase, respectively. Much higher Tg was also observed in the epoxy nanocomposites with the incorporation of DGO. 26 °C higher Tg are observed at 0.3 wt% DGO, compared to neat epoxy. Considering the low cost, high efficiency, and easy fabrication of modified GO nanoparticles, DGO/epoxy nanocomposites with enhanced mechanical properties will widen the application fields of epoxy resin. In addition, it will attract more research and industrial interests on such high performance epoxy composites.Acknowledgements
We are grateful for financial support by National Natural Science Foundation of China (51273118) and the Science & Technology Pillar Program of Sichuan (2013FZ0006), and the Analytical and Testing Center of Sichuan University for providing FTIR, AFM, Raman, TGA, SEM and DMA measurement.References
- A. Motahari, A. Omrani, A. A. Rostami and M. Ehsani, Thermochim. Acta, 2013, 574, 38–46 CrossRef CAS.
- D. Ratna, Handbook of thermoset resins, ISmithers Shawbury, UK, 2009 Search PubMed.
- T. Iijima, S. Miura, W. Fukuda and M. Tomoi, Eur. Polym. J., 1993, 29, 1103–1113 CrossRef CAS.
- M. E. Launey and R. O. Ritchie, Adv. Mater., 2009, 21, 2103–2110 CrossRef CAS.
- A. Yasmin, J. Luo, J. Abot and I. Daniel, Compos. Sci. Technol., 2006, 66, 2415–2422 CrossRef CAS.
- W. Boo, L. Sun, J. Liu, A. Clearfield, H.-J. Sue, M. Mullins and H. Pham, Compos. Sci. Technol., 2007, 67, 262–269 CrossRef CAS.
- W. Liu, S. V. Hoa and M. Pugh, Compos. Sci. Technol., 2005, 65, 2364–2373 CrossRef CAS.
- J. Qiu and S. Wang, J. Appl. Polym. Sci., 2011, 119, 3670–3674 CrossRef CAS.
- M. J. Palmeri, K. W. Putz and L. C. Brinson, ACS Nano, 2010, 4, 4256–4264 CrossRef CAS.
- M. Fang, Z. Zhang, J. Li, H. Zhang, H. Lu and Y. Yang, J. Mater. Chem., 2010, 20, 9635–9643 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- G. Tsoukleri, J. Parthenios, K. Papagelis, R. Jalil, A. C. Ferrari, A. K. Geim, K. S. Novoselov and C. Galiotis, Small, 2009, 5, 2397–2402 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- J. Ma, Q. Meng, A. Michelmore, N. Kawashima, Z. Izzuddin, C. Bengtsson and H.-C. Kuan, J. Mater. Chem. A, 2013, 1, 4255–4264 CAS.
- H. Yang, C. Shan, F. Li, Q. Zhang, D. Han and L. Niu, J. Mater. Chem., 2009, 19, 8856–8860 RSC.
- A. S. Wajid, H. Ahmed, S. Das, F. Irin, A. F. Jankowski and M. J. Green, Macromol. Mater. Eng., 2013, 298, 339–347 CrossRef CAS.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- K. A. Worsley, P. Ramesh, S. K. Mandal, S. Niyogi, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2007, 445, 51–56 CrossRef CAS.
- C. Zhang and T. Liu, Chin. Sci. Bull., 2012, 57, 3010–3021 CrossRef CAS.
- S.-Y. Yang, W.-N. Lin, Y.-L. Huang, H.-W. Tien, J.-Y. Wang, C.-C. M. Ma, S.-M. Li and Y.-S. Wang, Carbon, 2011, 49, 793–803 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS PubMed.
- L.-Z. Guan, Y.-J. Wan, L.-X. Gong, D. Yan, L.-C. Tang, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, J. Mater. Chem. A, 2014, 2, 15058–15069 CAS.
- Y.-J. Wan, L.-C. Tang, L.-X. Gong, D. Yan, Y.-B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Carbon, 2014, 69, 467–480 CrossRef CAS.
- I. Zaman, T. T. Phan, H.-C. Kuan, Q. Meng, L. T. B. La, L. Luong, O. Youssf and J. Ma, Polymer, 2011, 52, 1603–1611 CrossRef CAS.
- S. Chatterjee, J. Wang, W. Kuo, N. Tai, C. Salzmann, W. Li, R. Hollertz, F. Nüesch and B. Chu, Chem. Phys. Lett., 2012, 531, 6–10 CrossRef CAS.
- X. Li, H. Gao, W. A. Scrivens, D. Fei, X. Xu, M. A. Sutton, A. P. Reynolds and M. L. Myrick, Nanotechnology, 2004, 15, 1416 CrossRef CAS.
- J. Shen, W. Huang, L. Wu, Y. Hu and M. Ye, Compos. Sci. Technol., 2007, 67, 3041–3050 CrossRef CAS.
- Y. Hu, J. Shen, N. Li, H. Ma, M. Shi, B. Yan, W. Huang, W. Wang and M. Ye, Compos. Sci. Technol., 2010, 70, 2176–2182 CrossRef CAS.
- L. Li, Z. Zeng, H. Zou and M. Liang, Thermochim. Acta, 2015, 614, 76–84 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2010, 20, 1982–1992 RSC.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- J.-P. Tessonnier and M. A. Barteau, Langmuir, 2012, 28, 6691–6697 CrossRef CAS PubMed.
- M. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'Homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- C. Bao, Y. Guo, L. Song, Y. Kan, X. Qian and Y. Hu, J. Mater. Chem., 2011, 21, 13290–13298 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- J. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. Tascon, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- W.-H. Liao, S.-Y. Yang, J.-Y. Wang, H.-W. Tien, S.-T. Hsiao, Y.-S. Wang, S.-M. Li, C.-C. M. Ma and Y.-F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 869–877 CAS.
- J. Shen, Y. Hu, C. Li, C. Qin and M. Ye, Small, 2009, 5, 82–85 CrossRef CAS PubMed.
- T. Ramanathan, A. Abdala, S. Stankovich, D. Dikin, M. Herrera-Alonso, R. Piner, D. Adamson, H. Schniepp, X. Chen and R. Ruoff, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |