
Anthanthrene dye-sensitized solar cells: influence of the number of anchoring groups and substitution motif†
Abstract
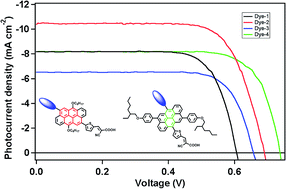
Four new dye molecules comprising an anthanthrene core, thiophene-cyanoacrylic acid electron acceptor and triphenylamine (TPA) electron donor units were prepared. Thereby, the substitution pattern at the anthanthrene core has been varied systematically. The molecules were fully characterized using optical spectroscopy and cyclic voltammetry, and their electronic structures and excitation energies were calculated by DFT and TDDFT methods. The photovoltaic properties of dye-sensitized solar cells based on these molecules were investigated. In comparison to symmetric dyes, the introduction of the organic donor TPA in asymmetric dyes leads to a remarkable improvement of the solar energy conversion efficiencies due to higher Voc and Jsc values of devices, giving rise to the best overall efficiency of 5.27%.
 Please wait while we load your content...
Please wait while we load your content...

