
An on-line spectroscopic monitoring system for the electrolytes in vanadium redox flow batteries
Abstract
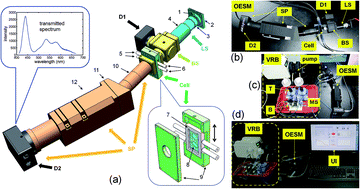
In this work, an on-line electrolyte spectroscopic monitoring system (OESM) is developed for long term monitoring of the electrolytes in a charge–discharge cycling VRB. We demonstrated experimentally that the transmittance spectra of the positive/negative electrolyte in a cycling VRB can be on-line monitored. With appropriate calibration, parameters such as the state of charge (SOC) of the electrolytes can be calculated. The system offers a tool for further research on the complex relationships between the spectra and the composition of the electrolyte in a VRB, and provides a dynamic method to study the kinetics of the electrolyte imbalance in VRBs.
 Please wait while we load your content...
Please wait while we load your content...

