
Methanol oxidation on Ru(0001) for direct methanol fuel cells: analysis of the competitive reaction mechanism†
Xiaoqing Lu*a,
Weili Wanga,
Zhigang Dengb,
Houyu Zhu*a,
Shuxian Weia,
Siu-Pang Ngb,
Wenyue Guo*a and
Chi-Man Lawrence Wub
aCollege of Science, China University of Petroleum, Qingdao, Shandong 266580, P. R. China. E-mail: luxq@upc.edu.cn; wyguo@upc.edu.cn; Fax: +86-532-86983363; Tel: +86-532-86983372
bDepartment of Physics and Materials Science, City University of Hong Kong, Hong Kong SAR, P. R. China
First published on 21st December 2015
Abstract
The competitive oxidation reaction mechanism of methanol on the Ru(0001) surface has been investigated by periodic density functional theory (DFT). Stable adsorption configurations, elementary reaction energies and barriers, the potential energy surface (PES), and the electrochemical potential analysis were elucidated. The results showed that O–H bond activation was more competitive than C–H and C–O bond activation during the initial methanol oxidation. Competitive pathways occurred for CH3OH oxidation to CH2O via CH3OH → CH3O → CH2O versus CH3OH → CH2OH → CH2O, further to COOH via the CO pathway CH2O → CHO → CO → COOH versus the non-CO pathway CH2O → CH2OOH → CHOOH → COOH, and finally oxidation to CO2. Taking PES and the electrochemical potential analysis into account, CH3OH → CH2OH → CH2O → CH2OOH → CHOOH → COOH → CO2 appeared to be the preferred oxidation pathway. The OH group could inhibit CO formation by directly reacting with CH2O to yield CH2OOH but could not efficiently remove the CO that had already been produced by the reactions.
1. Introduction
Direct methanol fuel cells (DMFCs) have attracted widespread attention as clean, portable, and high-efficiency energy conversion devices that utilize methanol (CH3OH) oxidation reactions on catalysts. A large number of investigations have been performed to explore the catalysis mechanisms of CH3OH on various pure metals, such as Ni,1,2 Cu,3,4 Ag,5 Pd,6–10 Pt,11–13 Rh,14,15 and Ru.16–19 Among these metals, Ru is generally applied as one of the main components of commercial catalysts in DMFC anodes, which can alleviate the problem of poor anode durability due to CO poisoning.20–24 To achieve rational modification on the anode catalysts, many efforts have been devoted to understanding the CH3OH oxidation mechanism on Ru catalysts experimentally. Unfortunately, although these studies were performed by using the same infrared absorption spectroscopy, conflicting evidence on adsorption and the reaction mechanism of CH3OH oxidation have been observed on clean16,17 and O-precovered25,26 Ru(0001) surfaces. For the adsorption of reaction intermediates, Barros et al.17 found upright and tilted methoxy (CH3O) and formaldehyde (CH2O); while Gazdzicki et al.19 proposed an upright configuration of CH3O. For CH3OH oxidation, Gazdzicki et al.18,19 found that CH3OH was adsorbed as an intact molecule on clean and O-precovered Ru(0001) at temperatures below 80 K, while CH3O was detected when annealing to 200–220 K; however, Barros et al.27 observed spontaneous O–H bond breakage to form CH3O for CH3OH at 80 K. Although these contradictions exist, researchers generally agreed that CH3OH could easily dehydrogenate on clean Ru(0001), leaving CO on its surface.16,17From the theoretical point of view, CH3OH oxidation on several clean metal surfaces has been investigated.2,8,10–12,28–33 On Ni(111),2 the favorable pathway of CH3OH decomposition involved initial O–H bond scission, followed by sequential hydrogen abstractions to achieve the final products CO and H via CH2O and CHO. On Pt(111),11,12 the C–H bond scission of CH3OH was the primary reaction process relative to the O–H bond scission, and the route CH3OH → CH2OH → CHOH → CO was suggested as the competitive pathway. On Pd(111), Schennach et al.8 and Jiang et al.10 found that initial C–H bond scission is preferable to that on Pt(111); by contrast, Zhang et al.6 compared initial O–H and C–O bond scissions processes and found that O–H bond scission was energetically favorable. Despite difference in the bond scission mechanisms involved, production CO on clean metal surfaces appeared to be unavoidable. When considering the electrochemical environment induced by introduction of the OH group, theoretical calculations showed that CO poisoning could be alleviated by further oxidizing to CO2 via CO + OH → COOH → CO2 + H, which is regarded as “the CO pathway”,28–30 and/or by “the non-CO pathway” along CHO + OH → CHOOH → CO2 + 2H.31–33 Although Ru has been widely used in commercial DMFC anodes, the CH3OH oxidation mechanism on Ru surfaces remains unclear because of contradictions observed in previous experiments and the lack of theoretical studies. Therefore, more details, including the most stable adsorption configuration, competitive reaction activation process, reaction thermodynamics and kinetics, and selectivity of production, are required to achieve a better understanding of CH3OH oxidation on Ru catalyst.
In this work, the competitive oxidation reaction mechanism of methanol on Ru(0001) surface was investigated by using periodic density functional theory (DFT). The Ru(0001) surface is selected because it is thermodynamically stable and commonly used as a model system.17,18 All possible adsorption structures and energies, elementary reaction steps, potential energy surface, and electrochemical potential analysis were elucidated along the competitive pathways from CH3OH to CH2OH, COOH, and CO2. The findings of this study are expected to provide deeper insights into the catalytic performance of Ru, adsorption selectivity, sequence of bond scission, competition of elementary reaction processes, thermodynamic and kinetic properties, and thus highlight the intrinsic characteristics of methanol oxidation on Ru(0001) in DMFCs.
2. Computational methods
All calculations were performed in the DFT framework with DMol3 in Materials Studio (Accelrys Inc.).34 The exchange-correlation energy was calculated within the generalized gradient approximation (GGA) using Perdew and Wang (PW91) functional.35,36 To take the relativity effect into account, the density functional semicore pseudopotential (DSPP)37,38 method was employed for Ru atoms, and C, H and O atoms were treated with the all-electron basis set. The valence electron functions were expanded into a set of numerical atomic orbitals by a double-numerical basis with polarization functions (DNP). Fermi smearing of 0.005 Hartree and a real-space cutoff of 4.7 Å were used to improve the computational performance.39,40 Transition state (TS) searches were performed at the same theoretical level by using the complete LST/QST method.41 The convergence criterion for the TS searches was set to 0.272 eV Å−1 for the root-mean-square of atomic forces. Vibrational frequencies were calculated for all the initial states (ISs) and final states (FSs), as well as the TSs from the Hessian matrix with harmonic approximation. Zero-point energy (ZPE) was calculated from the resulting frequencies.42Ru(0001) surface was modeled with a three-layer slab with nine Ru atoms per layer representing a (3 × 3) surface unit cell; a vacuum region of 12 Å thickness was used to separate the surface from its periodic image in the direction along the surface normal. The reciprocal space was sampled with a (4 × 4 × 2) k-point grid using the Monkhorst–Pack method.43 A single adsorbate was allowed to adsorb on one side of the unit cell, corresponding to a surface coverage of 1/9 ML. Full-geometry optimization was performed for relevant adsorbates and the uppermost two layers without symmetry constraints, while the bottom layer Ru atoms were fixed to their bulk-truncated positions with the experimentally determined lattice parameters of a = b = 2.71 Å and c = 4.28 Å. Nonperiodical structures were fully optimized at the same theoretical level for the isolated atoms, radicals and molecules involved.
The adsorption energies reported herein are calculated using the equation:
| Eads = Eadsorbate + EM − Eadsorbate/M | (1) |
3. Results and discussion
For clarity, this section is organized as follows: first, adsorption structures and energies for most of the intermediates are presented. Then, the possible elementary reactions along with the relevant reaction network are elucidated. Next, the overall PES for competitive pathways is discussed. Finally, the CH3OH electrooxidation process on Ru(0001) is analyzed. All energies reported herein are with ZPE corrections.3.1 Adsorption
Table 1 tabulates the adsorption information for the most stable adsorption modes of intermediates involved, and the corresponding configurations are shown in Fig. 1. Structural parameters and geometries of other intermediates are presented in Table S1 and Fig. S1 (ESI†).| Species | Site | Mode | Eads | dC/H–Ru | dO–Ru | Anglesa |
|---|---|---|---|---|---|---|
| a Values are angles between the surface normal and the C–O axis in the corresponding species. | ||||||
| CH3OH* | Top | η1(O) | 0.56 | 2.31 | 53 | |
| CH2OH* | Bridge | η1(C)–η1(O) | 1.98 | 2.14 | 2.28 | 90 |
| CH3O* | fcc | η3(O) | 2.52 | 2.20, 2.20, 2.20 | 0 | |
| hcp | η3(O) | 2.54 | 2.19, 2.19, 2.19 | 0 | ||
| CH2O* | Top | η1(O) | 0.45 | 2.20 | 49 | |
| fcc | η1(C)–η2(O) | 0.90 | 2.15 | 2.20, 2.20 | 79 | |
| hcp | η1(C)–η2(O) | 0.96 | 2.15 | 2.18, 2.18 | 79 | |
| CHO* | Bridge | η1(C)–η1(O) | 2.44 | 1.99 | 2.19 | 81 |
| fcc | η2(C)–η1(O) | 2.26 | 2.12 | 2.15, 2.19 | 76 | |
| hcp | η2(C)–η1(O) | 2.36 | 2.11 | 2.13, 2.17 | 76 | |
| CO* | Top | η1(C) | 1.93 | 1.89 | 0 | |
| Bridge | η2(C) | 1.66 | 2.08, 2.08 | 0 | ||
| fcc | η3(C) | 1.70 | 2.15, 2.16, 2.17 | 0 | ||
| hcp | η3(C) | 1.71 | 2.14, 2.14, 2.15 | 0 | ||
| CH2OOH* | fcc | η1(O)–η2(O) | 2.73 | 2.24, 2.22, 2.22 | 42, 67 | |
| hcp | η1(O)–η2(O) | 2.77 | 2.32, 2.19, 2.21 | 43, 72 | ||
| CHOOH* | fcc | η1(C)–η2(O) | 0.62 | 2.19 | 2.10, 2.28 | 87, 90 |
| hcp | η1(C)–η2(O) | 0.66 | 2.18 | 2.09, 2.27 | 88, 90 | |
| trans-COOH* | Bridge | η1(C)–η1(O) | 2.69 | 2.05 | 2.20 | 33, 83 |
| cis-COOH* | Bridge | η1(C)–η1(O) | 2.68 | 2.05 | 2.21 | 31, 85 |
| CO2* | Bridge | η1(C)–η1(O) | 0.05 | 2.10 | 2.22 | 59, 79 |
| OH* | Top | η1(O) | 2.84 | 1.99 | ||
| Bridge | η2(O) | 3.14 | 2.16, 2.16 | |||
| fcc | η3(O) | 3.19 | 2.21, 2.21, 2.21 | |||
| hcp | η3(O) | 3.19 | 2.20, 2.20, 2.20 | |||
| H* | Bridge | η2(H) | 2.80 | 1.82, 1.82 | ||
| fcc | η3(H) | 2.91 | 1.90, 1.91, 1.92 | |||
| hcp | η3(H) | 2.87 | 1.90, 1.91, 1.92 | |||
 | ||
| Fig. 1 The most stable adsorption configurations of intermediates involved in CH3OH oxidation on Ru(0001). The C, O, H, and Ru atoms are shown in gray, red, white, and light-blue colors, respectively. | ||
3.2 Initial oxidation of CH3OH to CH2O
The reaction network of CH3OH oxidation on Ru(0001) is shown in Fig. 2. Structures of IS, TS, and FS along elementary reaction steps are presented in Fig. 3–6. As a whole, CH3OH dehydrogenates to CH2O via the competitive routes of CH3OH → CH3O → CH2O and/or CH3OH → CH2OH → CH2O (Fig. 3), then further oxidizes to trans-COOH via the CO pathway CH2O → CHO → CO → COOH (Fig. 4) and/or the non-CO pathway CH2O → CH2OOH → CHOOH → COOH (Fig. 5), and achieve CO2 eventually (Fig. 6). For brevity, other elementary reaction steps with relatively high energy barriers are described in Fig. S2,† including the C–O bond scissions of CH3OH, CH3O, CH2OH, CH2O, and CHO, the combination of CHO and OH to CHOOH, the reaction of CO with OH to cis-COOH, the dehydrogenation of CH2OOH to CH2OO, and further dehydrogenation to CHOO. | ||
| Fig. 2 The reaction network for CH3OH oxidation on Ru(0001). “A–B” represents the “A–B” bond scission and “+OH” means the combination with OH group. The energy barrier and reaction energy (in parentheses) are in eV. For simplicity, the H atom is omitted. | ||
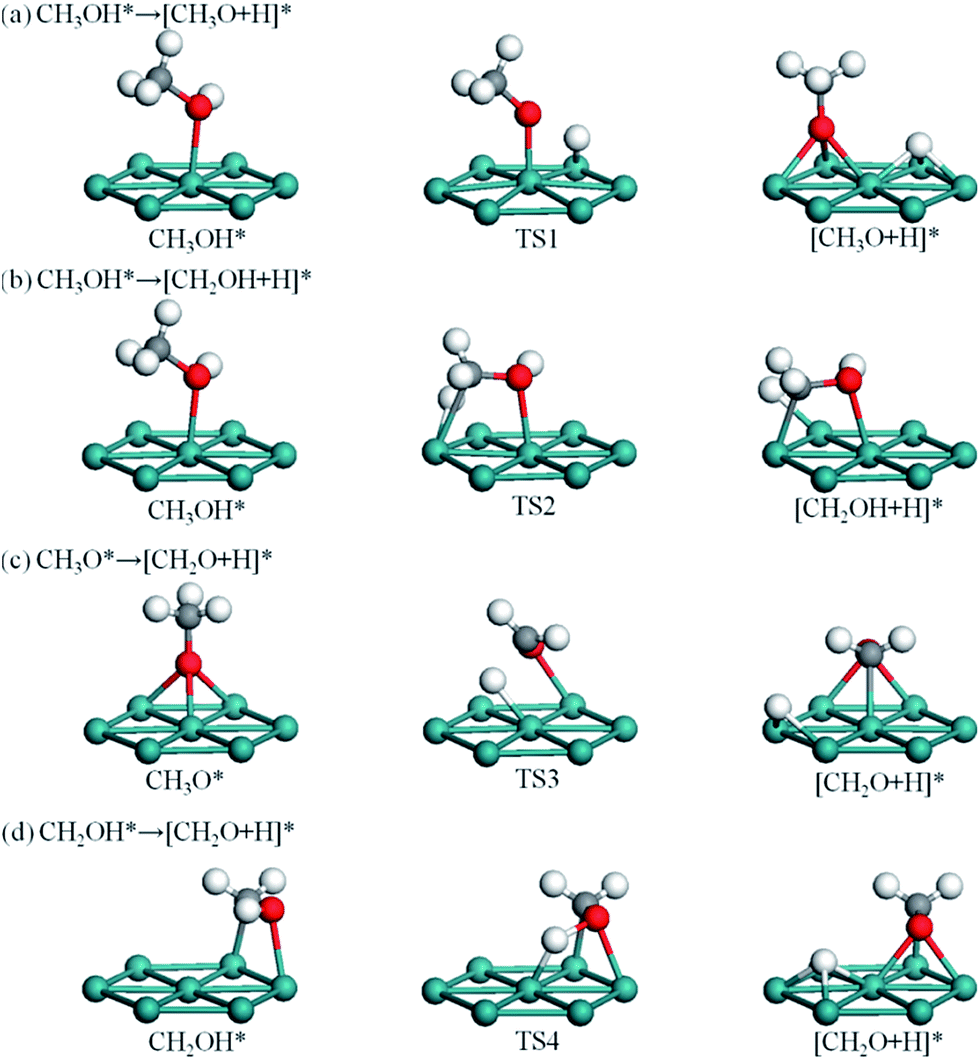 | ||
| Fig. 3 Dehydrogenation of CH3OH to CH2O through CH3OH → CH3O → CH2O (a and c) and CH3OH → CH2OH → CH2O (b and d). [A + B]* denotes the coadsorbed A and B species. | ||
 | ||
| Fig. 4 The CO pathway for CH2O oxidation to trans-COOH. | ||
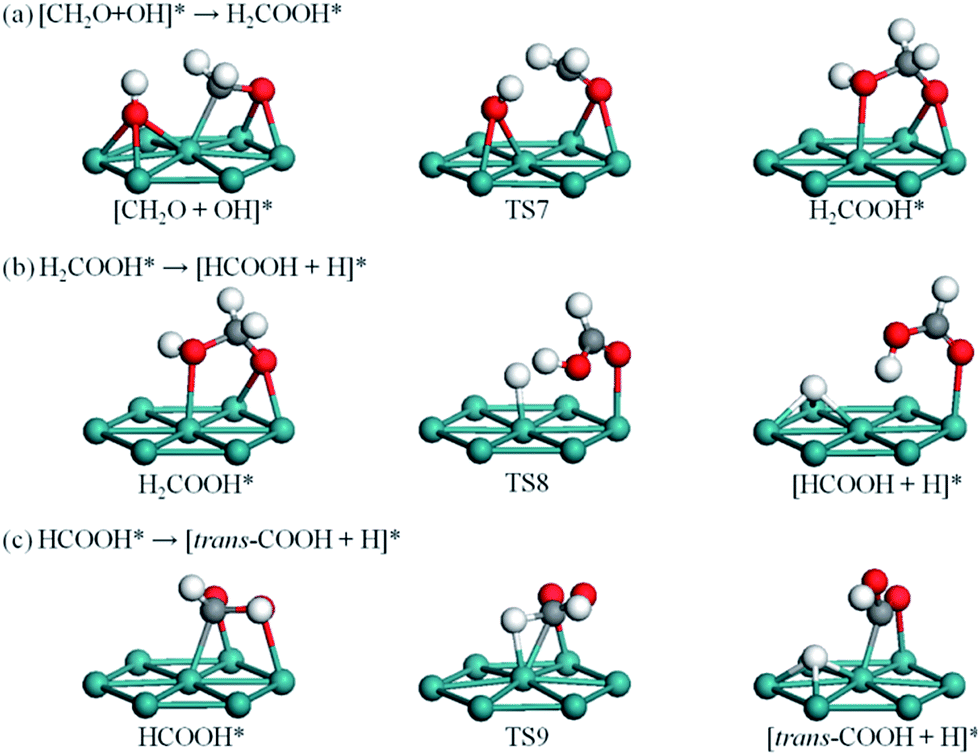 | ||
| Fig. 5 The non-CO pathway for CH2O oxidation to trans-COOH. | ||
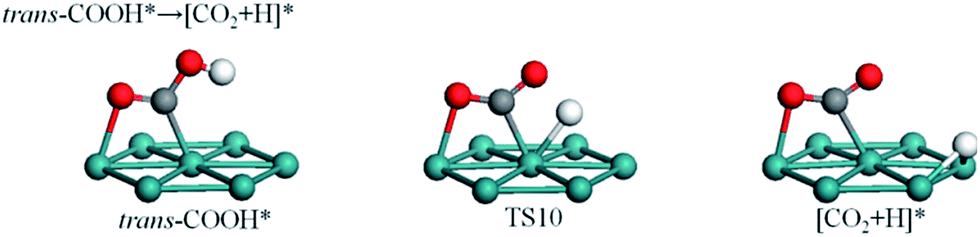 | ||
| Fig. 6 Formation of CO2 via dehydrogenation of trans-COOH. | ||
Three possible initial bond scissions of CH3OH are considered, that is, O–H, C–H and C–O bond scissions. C–O bond scission involves a very high energy barrier of 1.68 eV, whereas O–H bond scission involves a relatively low energy barrier of 0.56 eV. The reaction CH3OH → CH3O + H leads to elongation of the O–H bond length to 2.10 Å in TS1 (Fig. 3a) relative to 0.98 Å in IS and yields coadsorbed CH3O and H atom on adjacent hcp sites as FS. As shown in Fig. 3b, C–H bond scission is mirrored by an elongated C–H bond length of 1.58 Å in TS2, together with the CH2OH group bending towards the surface and the H atom moving to adjacent bridge site. The energy barrier of CH3OH → CH2OH + H is 0.83 eV. The reaction process of CH3OH → CH3 + OH is presented in Fig. S2.† The activation energies of the C–H and C–O bonds are higher than that of the O–H bond, indicating that initial O–H bond scission of CH3OH is highly favorable on Ru(0001). During initial oxidation of CH3OH, O–H bond scission is also preferable on Cu(111)52 and Ag(111),5 while C–H bond scission is more facile on Pd(111) and Pt(111).53,54
Subsequently, C–H bond scission of CH3O starts with its most stable configuration at hcp site and ends with CH2O at hcp site and H atom at fcc site, as shown in Fig. 3c. In TS3, the C–O axis tilts towards the surface with the O atom above bridge site, thereby facilitating the C–H bond scission. The energy barrier is calculated to be 1.01 eV and the reaction is exothermic by 0.07 eV. For CH2OH, C–H bond scission involves a high energy barrier of 1.07 eV relative to the 0.76 eV of its O–H bond scission. As shown in Fig. 3d, O–H bond scission of CH2OH yields CH2O at fcc site and the H atom at hcp site. The O–H bond length is elongated to 1.48 Å in TS4, and a slight swag vibration facilitates removal of the H atom.
3.3 CH2O oxidation to COOH via the CO pathway
As shown in Fig. 4a, dehydrogenation of CH2O to CHO involves rotation of CH2O along the C–O bond axis, so that one of the H atoms can easily approach a surface Ru atom and form a Ru–H bond. This process is exothermic by 0.48 eV and the energy barrier is 0.53 eV. By comparison, C–O bond scission requires a higher energy barrier of 1.26 eV, as shown in Fig. S2c and Table S1.†Fig. 4b illustrates that dehydrogenation of CHO starts with CHO binding at bridge site in IS and ends with CO sitting atop a Ru atom in FS. In TS5, CHO is located at off-top site and the C–H bond is elongated by 0.10 Å; C–H bond scission is triggered by a swag vibration of the adsorbed CHO, giving rise to the H atom close to the surface. This process is strongly exothermic by 1.17 eV with a relatively low energy barrier of 0.34 eV. Comparatively, C–O bond scission and combination of CHO with OH to form CHOOH involve energy barriers of up to 1.62 and 1.92 eV, respectively, as shown in Fig. S2c, d and Table S1.†
Oxidation of the adsorbed CO by OH can produce trans-COOH and cis-COOH in FS. The energy barriers for these reaction steps are as high as 1.74 and 2.07 eV, respectively. To achieve the formation of trans-COOH, as shown in Fig. 4c, CO and OH groups approach each other together with the elevation of the OH group in TS6.
3.4 CH2O oxidation to COOH via the non-CO pathway
CH2O can react with OH to form CH2OOH on Pd,53 Ag,54,55 and Cu.56 In the real electrochemical environment of DMFCs, the OH group usually comes from the reaction between coadsorbed H2O and Oad (H2O + Oad → 2OH)57 or direct decomposition of H2O (H2O → H + OH). On Ru(0001), the adsorbed CH2O at hcp site reacts with OH at fcc site in IS and ends with CH2OOH at hcp site, as shown in Fig. 5a. The binding energy for coadsorbed CH2O and OH is 5.68 eV, much higher than the sum of their individual adsorption energies of 0.96 eV for CH2O and 3.19 eV for OH. This result indicates the strong attraction between CH2O and OH, which stabilizes the coadsorption system so as to further bind these two species. In TS7, the OH group migrates to bridge site, thus facilitating interactions with the elevated CH2 group in CH2O. The energy barrier here is 0.97 eV, and the reaction is exothermic by 0.13 eV.As shown in Fig. 5b, CH2OOH dehydrogenation via C–H bond scission results in CHOOH located at top site and the H atom at hcp site. In TS8, the C–H bond is broken and the detached H atom sits at top site. This process is exothermic by 0.63 eV, and the energy barrier is 0.24 eV. CH2OOH dehydrogenation can also proceed via O–H bond scission (Fig. S2g†), that is, CH2OOH → CH2OO + H. The barrier here is 0.92 eV and the reaction energy is 0.18 eV.
Fig. 5c illustrates C–H bond scission of CHOOH, which starts with CHOOH located at hcp site and ends with trans-COOH at bridge site and H atom at fcc site. In TS9, the C–H bond distance is elongated by 0.21 Å with the OH group detached from the surface; slight rotation of the C–O bond facilitates the C–H bond scission. This dehydrogenation step is exothermic by 0.72 eV, and the energy barrier is 0.38 eV. O–H bond scission for CHOOH (CHOOH → CHOO + H, Fig. S2h†) involves an energy barrier of 0.61 eV and a reaction energy of 0.54 eV.
3.5 Formation of CO2
Fig. 6 reveals that stretch vibrations of the O–H bond can induce the H atom of trans-COOH to approach the surface such that trans-COOH decomposes to COO and atomic H. Obviously, O–H bond scission is more likely to take advantage of the downward-facing mode of trans-COOH than the up-facing mode of cis-COOH. In TS10, the CO2 fragment remains at bridge site while the O–H bond is broken with an O–H distance of 1.53 Å. This process is almost thermoneutral with a reaction energy of 0.01 eV and an energy barrier of 0.96 eV.3.6 Potential energy surface (PES)
The overall PES of CH3OH oxidation on Ru(0001) is illustrated in Fig. 7, including three-stage oxidation products of CH2O, COOH and CO2. Competitive pathways appear for the first two steps, that is, CH3OH → CH3O → CH2O versus CH3OH → CH2OH → CH2O for CH2O; and the CO pathway CH2O → CHO → CO → trans-COOH versus the non-CO pathway CH2O → CH2OOH → CHOOH → trans-COOH for COOH. | ||
| Fig. 7 PES of CH3OH oxidation on Ru(0001). All energies (eV) are relative to the energy gaseous molecules of hydroxy and one gaseous molecule of methanol plus the clean Ru(0001) surface with ZPE corrections. [A + B]* denotes the coadsorbed A and B, and A* + B* represents respective adsorptions of A and B on two separated slabs. | ||
For CH3OH oxidation to CH2O, the dehydrogenation sequence plays a crucial role in the reaction kinetics via two competitive pathways. Along the CH3OH → CH3O → CH2O pathway, the activation energies of O–H and C–H bond scission are 0.56 and 1.01 eV, respectively. By comparison, along the CH3OH → CH2OH → CH2O pathway, the energy barriers of C–H and O–H bond scissions are 0.83 and 0.76 eV, respectively. Thus, to achieve oxidation of CH3OH on Ru(0001), initial O–H bond scission (Ea = 0.56 eV) is more facile than the C–H bond scission (Ea = 0.83 eV). Once CH3O is formed, subsequent C–H bond scission involves a larger barrier of 1.01 eV. Therefore, CH3O can accumulate to a certain extent on the Ru surface, which explains why CH3O is consistently detected in the experiments.18,19 Considering the relative smooth PES, the CH3OH → CH2OH → CH2O pathway is more competitive for forming CH2O than the CH3OH → CH3O → CH2O pathway.
For CH2O oxidation to COOH, the energy barriers along the CO pathway are 0.53 eV for CH2O → CHO, 0.34 eV for CHO → CO, and 1.74 eV for CO + OH → COOH. The rate-determining step along the CO pathway appears to be the combination of CO and co-adsorbed OH to form COOH. By comparison, the energy barriers along the non-CO pathway are 0.97 eV for CH2O + OH → CH2OOH, 0.24 eV for CH2OOH → CHOOH + H, and 0.38 eV for CHOOH → COOH + H. The rate-determining step along the non-CO pathway appears to be the combination of CH2O and co-adsorbed OH to produce CH2OOH. Note that the rate-determining steps along both pathways appear when reacting with the coadsorbed OH to form the C–O bond. Comparing these two pathways, CO seems to be easier to form along the CO pathway and accumulate on Ru(0001) because of the high energy barrier required for further combination with OH. However, since the OH group pre-exists in the electrolyte and has a much larger adsorption energy than CO (2.84–3.19 eV for OH versus 1.66–1.93 eV for CO, Table 1), most of the adsorption sites would be covered by OH under working conditions. Thus, the coverage of newly-formed CO is low and the formation of CO on Ru(0001) is inhibited. Comparatively, the non-CO pathway is more favorable and followed by COOH dehydrogenization to form CO2. Participation of the OH group on Ru(0001) avoid the CO formation during CH3OH oxidation along the non-CO pathway, but cannot remove the CO that had already been produced because of the very high barriers involved in the transformation of CO + OH → COOH (1.74 eV for trans-COOH and 2.07 eV for cis-COOH).
3.7 Electrochemical potential analysis
The standard hydrogen electrode (SHE)58 is used to treat the electrochemical potential (U). The overall electrochemical reaction on the anode of a DMFC is: CH3OH + H2O → CO2 + 6H+ + 6e−. Under standard conditions, the free energy of two protons and two electrons at zero potential is equal to the free energy of a hydrogen molecule. The change in free energy at a given potential U of a reaction involving the formation of a proton electron pair versus that at the SHE is equivalent to ΔG = −eU, where e is the charge on the electron. Additionally, intermediates that are closed-shell molecules (e.g., CH3OH and CO2) are treated as gas-phase molecules, as the bond energy to the surface will be unlikely to overcome the entropy loss through surface binding. Based on this approach, the required lowest potential U can be obtained to discuss the competitive reaction pathways of CH3OH electro-oxidation on Ru(0001), as successfully applied in similar reactions.54,58–60 The details of this method refer to the previous literature.54,59The free energies of all of the intermediates are calculated relative to H2O(l), CO2(g) and H2(g), as presented in Table 2. The free energy of CH3OH, for example, is calculated as follows,
| CO2(g) + 3H2(g) → CH3OH(g) + H2O(l) | (2) |
| CH3OH | CH3O | CH2OH | CH2O | H2COOH | CHO | CHOOH | CO | COOH | CO2 |
|---|---|---|---|---|---|---|---|---|---|
| a CO2(g), H2O(l), and H2(g) are used as reference. Zero-point energy and entropy corrections are included. | |||||||||
| 0.10 | −0.01 | 0.32 | 0.63 | 0.36 | 0.16 | 0.28 | −0.79 | −0.05 | 0.00 |
ΔGCH3OH = TEH2O + TECH3OH − TECO2 − 3TEH2 + ZPEH2O + ZPECH3OH − ZPECO2 − 3ZPEH2 − T(SH2O + SCH3OH − 3SH2 − SCO2), where TE is the total energy of the reactant and product species, T is the standard temperature (298 K), ZPE is the zero-point energy, and S is the entropy of the species. For gas- and liquid-phase molecules, the entropy values are taken from the literature.61 The free energies of surface intermediates are also calculated in a similar manner, for example,
| CO2(g) + 2.5H2(g) + * → CH3O* + H2O(l) | (3) |
ΔGCH3O* = TEH2O + TECH3O* − TECO2 − 2TEH2 − TEclean + ZPEH2O + ZPECH3O* − ZPECO2 − 2.5ZPEH2 − T(SH2O + SCH3O* − 2.5SH2 − SCO2), where TEclean is the total energy of the clean slab, TECH3O* is the total energy of CH3O* adsorbed on a clean slab, and ZPECH3O* and SCH3O* are the zero-point energy and entropy for the adsorbed CH3O*, respectively. For molecules bound to the surface, the vibrational entropy is calculated assuming a quantum mechanical harmonic oscillator with the same vibrational frequencies as those applied to the zero-point energy. As shown in Table 2, the free energies for closed-shell gas-phase intermediates are as follows: CH3OH = 0.10 eV, CH2O = 0.63 eV, CHOOH = 0.28 eV, and CO2 = 0.0 eV; these values are comparable with previous DFT results54 of 0.11, 0.71 eV, 0.42 eV, and 0.0 eV, respectively. As a reference, the standard table values are: CH3OH(g) = 0.05 eV, CH2O(g) = 0.57 eV, CHOOH(g) = 0.44 eV, and CO(g) = 0.0 eV. The negative values of adsorbed CH3O, CO, and COOH indicate that these surface intermediates are thermodynamically more stable than the reference molecules at standard conditions [i.e., CO2(g), H2O(l), and H2(g) at 298 K and 1 bar pressure]. Of these intermediates, CO has the lowest free energy because of its strong bonding with Ru(0001), as noted previously. Positive values of all other adsorbed intermediates (i.e., CH2OH, CH2OOH, and CHO) suggest that the surface species is not as stable as the reference molecules at standard conditions; thus, the reaction to form the reference molecules from the surface species would be exothermic on Ru(0001) at standard conditions.
A comparison of the electrochemical potentials for the competitive reactions on Ru(0001) is shown in Fig. 8. For the initial CH3OH oxidation processes CH3OH → CH3O versus CH3OH → CH2OH, a lower potential is required to strip off the hydroxyl proton than that required to strip off the methoxyl proton, in agreement with previous experimental observations.62 Similar case takes place for proton stripped from the hydroxyl with a lower potential along the subsequent oxidation to formaldehyde (CH2OH → CH2O versus CH3O → CH2O). Along the CO pathway CH2O → CHO → CO → COOH, the highest potential is calculated to be 0.74 V for CO + OH → COOH; by contrast, the non-CO pathway CH2O → CH2OOH → CHOOH → COOH requires a much lower potential than 0.74 V. Therefore, at low potentials, CH3OH electrooxidation on Ru(0001) proceeds primarily via the non-CO pathway to produce CO2; at high potentials, both the CO and non-CO pathways are thermodynamically available on Ru(0001).
 | ||
| Fig. 8 The electrochemical potentials for CH3OH oxidation on Ru(0001). The x-axis indicates how many proton/electron pairs have been created from CH3OH. | ||
4. Conclusions
Our theoretical investigation provides a systematic understanding of the competitive oxidation reaction mechanism of methanol on Ru(0001) surface for DMFCs. The following findings are obtained:(1) To achieve a stable adsorption configuration, CH3OH and CO prefer to adsorb at top site, CH2OH, CHO and COOH prefer to adsorb at bridge site, and CH3O, CH2O, CH2OOH and CHOOH prefer to adsorb at hollow site on the Ru(0001) surface.
(2) O–H bond activation is more competitive than C–H and C–O bonds activations during CH3OH and CH2OH oxidation. Competitive oxidation pathways to CH2O occur via CH3OH → CH3O → CH2O versus CH3OH → CH2OH → CH2O, further to COOH by virtue of OH group via the CO pathway CH2O → CHO → CO + OH → COOH versus the non-CO pathway CH2O + OH → CH2OOH → CHOOH → COOH, and finally oxidation to CO2. For both the CO and non-CO pathways, the rate-determining step appears to be the C–O bond formation when interacting with the OH group.
(3) PES analysis confirms that CH3OH → CH2OH → CH2O → CH2OOH → CHOOH → COOH → CO2 is the preferred pathway, agreeing well with the electrochemical potential analysis for CH3OH electro-oxidation. The OH group could inhibit CO formation by directly reacting with CH2O to yield CH2OOH, but cannot remove the CO that had already been produced.
Acknowledgements
This work was supported by NSFC (21303266), National Basic Research Program of China (2014CB239204), Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province (BS2013CL031), and the Fundamental Research Funds for the Central Universities (15CX05050A, 15CX08010A, 14CX02214A).Notes and references
- Y. H. Zhou, P. H. Lv and G. C. Wang, J. Mol. Catal. A: Chem., 2006, 258, 203–215 CrossRef CAS.
- G. C. Wang, Y. H. Zhou, Y. Morikawa, J. Nakamura, Z. S. Cai and X. Z. Zhao, J. Phys. Chem. B, 2005, 109, 12431–12442 CrossRef CAS PubMed.
- S. Sakong and A. Groß, J. Catal., 2005, 231, 420–429 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Catal., 2002, 208, 291–300 CrossRef CAS.
- W. S. Sim, P. Gardner and D. A. King, J. Phys. Chem., 1995, 99, 16002–16010 CrossRef CAS.
- C. J. Zhang and P. Hu, J. Chem. Phys., 2001, 115, 7182–7186 CrossRef CAS.
- K. H. Lim, Z. X. Chen, K. M. Neyman and N. Rösch, J. Phys. Chem. B, 2006, 110, 14890–14897 CrossRef CAS PubMed.
- R. Schennach, A. Eichler and K. D. Rendulic, J. Phys. Chem. B, 2003, 107, 2552–2558 CrossRef CAS.
- Z. X. Chen, K. M. Neyman, K. H. Lim and N. Rösch, Langmuir, 2004, 20, 8068–8077 CrossRef CAS PubMed.
- R. B. Jiang, W. Y. Guo, M. Li, D. L. Fu and H. H. Shan, J. Phys. Chem. C, 2009, 113, 4188–4197 CAS.
- S. K. Desai, M. Neurock and K. Kourtakis, J. Phys. Chem. B, 2002, 106, 2559–2568 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 3910–3919 CrossRef CAS PubMed.
- M. T. Koper, T. E. Shubina and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 686–692 CrossRef CAS.
- Y. Li and M. Bowker, Catal. Today, 1991, 10, 421–424 CrossRef CAS.
- R. B. Jiang, W. Y. Guo, M. Li, H. Y. Zhu, L. M. Zhao, X. Q. Lu and H. H. Shan, J. Mol. Catal. A: Chem., 2011, 344, 99–110 CrossRef CAS.
- A. A. Deckert, J. L. Brand, C. H. Mak, B. G. Koehler and S. M. George, J. Chem. Phys., 1987, 87, 1936–1947 CrossRef CAS.
- R. B. Barros, A. R. Garcia and L. M. Ilharco, J. Phys. Chem. B, 2001, 105, 11186–11193 CrossRef CAS.
- P. Gazdzicki, P. Uvdal and P. Jakob, J. Chem. Phys., 2009, 130, 224703 CrossRef PubMed.
- P. Gazdzicki and P. Jakob, J. Phys. Chem. C, 2010, 114, 2655–2663 CAS.
- G. Q. Lu and A. Wieckowski, Curr. Opin. Colloid Interface Sci., 2000, 5, 95–100 CrossRef CAS.
- U. A. Paulus, U. Endruschat, G. J. Feldmeyer, T. J. Schmidt, H. Bönnemann and R. J. Behm, J. Catal., 2000, 195, 383–393 CrossRef CAS.
- E. Antolini, Mater. Chem. Phys., 2003, 78, 563–573 CrossRef CAS.
- Q. Y. Lu, B. Yang, L. Zhuang and J. T. Lu, J. Phys. Chem. B, 2005, 109, 1715–1722 CrossRef CAS PubMed.
- H. S. Liu, C. J. Song, L. Zhang, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- R. B. Barros, A. R. Garcia and L. M. Ilharco, J. Phys. Chem. B, 2004, 108, 4831–4839 CrossRef CAS.
- R. B. Barros, A. R. Garcia and L. M. Ilharco, ChemPhysChem, 2005, 6, 1299–1306 CrossRef CAS PubMed.
- R. Brito de Barros, A. R. Garcia and L. M. Ilharco, Surf. Sci., 2003, 532–535, 185–190 CrossRef CAS.
- J. S. Spendelow, J. D. Goodpaster, P. J. A. Kenis and A. Wieckowski, J. Phys. Chem. B, 2006, 110, 9545–9555 CrossRef CAS PubMed.
- X. Q. Gong, P. Hu and R. Raval, J. Chem. Phys., 2003, 119, 6324–6334 CrossRef CAS.
- E. A. Batista, T. Iwasita and W. Vielstich, J. Phys. Chem. B, 2004, 108, 14216–14222 CrossRef CAS.
- M. Yin, Y. J. Huang, L. Liang, J. H. Liao, C. P. Liu and W. Xing, Chem. Commun., 2011, 47, 8172–8174 RSC.
- D. W. Yuan, X. G. Gong and R. Q. Wu, J. Chem. Phys., 2008, 128, 064706 CrossRef PubMed.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792–5798 CrossRef CAS.
- B. Delley, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- X. Q. Lu, Z. G. Deng, K. S. Chau, L. F. Li, Z. Q. Wen, W. Y. Guo and C. M. L. Wu, ChemCatChem, 2013, 5, 1832–1841 CrossRef CAS.
- Z. G. Deng, X. Q. Lu, Z. Q. Wen, S. X. Wei, Y. J. Liu, D. L. Fu, L. M. Zhao and W. Y. Guo, Phys. Chem. Chem. Phys., 2013, 15, 16172–16182 RSC.
- L. M. Zhao, S. P. Wang, Q. Y. Ding, W. B. Xu, P. P. Sang, Y. H. Chi, X. Q. Lu and W. Y. Guo, J. Phys. Chem. C, 2015, 119, 20389–20400 CAS.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- X. Q. Lu, Z. G. Deng, S. X. Wei, Q. Y. Zhu, W. L. Wang, W. Y. Guo and C. M. L. Wu, Catal. Sci. Technol., 2015, 5, 3246–3258 CAS.
- D. J. Chadi, Phys. Rev. B: Solid State, 1977, 16, 1746–1747 CrossRef.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- M. Mavrikakis and M. A. Barteau, J. Mol. Catal. A: Chem., 1998, 131, 135–147 CrossRef CAS.
- B. A. Sexton and A. E. Hughes, Surf. Sci., 1984, 140, 227–248 CrossRef CAS.
- J. Miragliotta, R. S. Polizzotti, P. Rabinowitz, S. D. Cameron and R. B. Hall, Chem. Phys., 1990, 143, 123–130 CrossRef CAS.
- M. N. D. S. Cordeiro, A. S. S. Pinto and J. A. N. F. Gomes, Surf. Sci., 2007, 601, 2473–2485 CrossRef CAS.
- R. Shekhar, M. A. Barteau, R. V. Plank and J. M. Vohs, J. Phys. Chem. B, 1997, 101, 7939–7951 CrossRef CAS.
- M. A. Barteau, J. Q. Broughton and D. Menzel, Surf. Sci., 1983, 133, 443–452 CrossRef CAS.
- M. Y. Chou and J. R. Chelikowsky, Phys. Rev. Lett., 1987, 59, 1737–1740 CrossRef CAS PubMed.
- A. V. de Carvalho, M. C. Asensio and D. P. Woodruff, Surf. Sci., 1992, 273, 381–384 CrossRef CAS.
- X. K. Gu and W. X. Li, J. Phys. Chem. C, 2010, 114, 21539–21547 CAS.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS PubMed.
- P. Ferrin, A. U. Nilekar, J. Greeley, M. Mavrikakis and J. Rossmeisl, Surf. Sci., 2008, 602, 3424–3431 CrossRef CAS.
- S. Lin, R. S. Johnson, G. K. Smith, D. Xie and H. Guo, Phys. Chem. Chem. Phys., 2011, 13, 9622–9631 RSC.
- M. J. T. C. van der Niet, A. den Dunnen, L. B. F. Juurlink and M. T. M. Koper, Angew. Chem., Int. Ed., 2010, 49, 6572–6575 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- G. S. Karlberg, J. Rossmeisl and J. K. Norskov, Phys. Chem. Chem. Phys., 2007, 9, 5158–5161 RSC.
- CRC Handbook of Chemistry and Physics, CRC Press, New York, 1996 Search PubMed.
- P. Gazdzicki and P. Jakob, J. Phys. Chem. C, 2010, 114, 2655–2663 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The metastable adsorption configurations, and other elementary reaction steps with relatively high energy barriers in CH3OH oxidation routes are presented. See DOI: 10.1039/c5ra21793h |
| This journal is © The Royal Society of Chemistry 2016 |