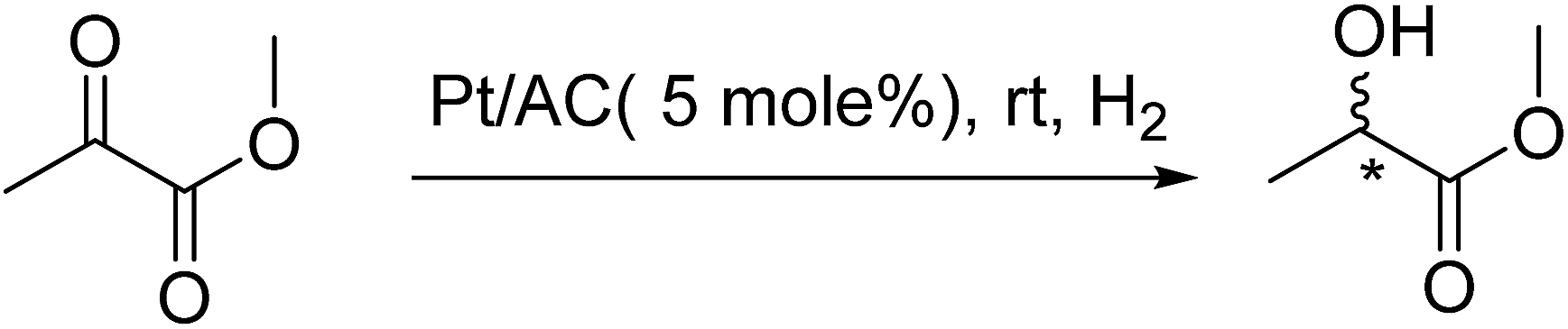DOI:
10.1039/C5RA21790C
(Paper)
RSC Adv., 2015,
5, 102481-102487
Platinum functionalized multiwall carbon nanotube composites as recyclable catalyst for highly efficient asymmetric hydrogenation of methyl pyruvate†
Received
19th October 2015
, Accepted 23rd November 2015
First published on 24th November 2015
Abstract
Platinum functionalized carbon materials such as carbon fibres, graphene, MWNTs (multiwalled carbon nanotubes) and activated carbon were used as heterogeneous catalytic systems for asymmetric hydrogenation of α-ketoester i.e. methyl pyruvate using cinchonidine (CD) as a chiral modifier. Interestingly, the MWNTs exhibited excellent enantioselectivity (>99% ee) and conversion (99%) in comparison to other Pt/C systems due to their high surface area. Furthermore, in the case of Pt/MWNTs, Pt nanoparticles are found to be uniformly dispersed and bound to the MWNTs acting like a single atom catalyst. Time-dependent nuclear magnetic resonance (NMR) studies, cyclic voltammetry (CV) and diffuse reflectance spectroscopy (DRS) have been carried out to study substrate–modifier–catalyst interactions. Recyclability of the catalyst was also tested up to ten cycles without losing any significant catalytic activity.
Introduction
Metal nanoparticle/carbon composite (M/C) systems are of great interest as they blend the exclusive properties of carbon materials with metal nanoparticles.1 Carbon based materials such as MWNTs, carbon fibers (CF), activated carbon (AC) and graphene are some of the key nominees due to their commercial availability and ease of chemical modification. MWNTs and graphene particularly prove to be an excellent choice due to their extraordinary properties including high specific surface area, electrical conductivity, good chemical and thermal stability etc.2–5 These materials can be used as composites with other materials or loaded with metals via chemical deposition. Since pristine carbon materials are chemically inactive, chemical tuning for the attachment of metal nanoparticles for catalytic applications is essential.6 These composites are used in variety of applications, as an example, as counter electrode in dye sensitized solar cell (DSSC),7 dehydrogenation of ammonia borane8 and as an electrocatalyst support for direct methanol fuel cells.9 M/C composites form a highly stable system that is unreactive in many acid and basic media, which do not promote any side reactions.10 The M/C systems are believed to serve as nanoreactors for catalytic conversion.11
Asymmetric homogenous catalysts have been widely used in various organic transformations due to their high reactivity and enantioselectivity.12 Homogeneous catalysis has an advantage of molecular reaction phenomena and ease of characterization of the reaction intermediates. Despite various advantages, separation and purification of product as well as recycling of the catalysts make it less environmentally benign.13–19 Strategic development of asymmetric heterogeneous catalysts is a viable option to overcome such issues by offering innate practical advantages over homogeneous catalyst.20
Various studies on asymmetric hydrogenation incorporated the use of conventional supporting materials such as Pt loaded on alumina (Al2O3), silica (SiO2) and titanium oxide (TiO2).20–25 The Orito reaction where chirally modified Pt/Al2O3 has seen a resurgence of activity because of its synthetic use,26,34 and considered as standard catalyst for asymmetric hydrogenation of α-functionalized carbonyls.26–35 The chirally modified commercial Pt/Al2O3 catalyst has been used extensively for wide range of carbonyl substrates with high optical and chemical conversion.33 Impressive results with ee's higher than 95% were obtained with synthetically modified nanoparticulated platinum supported alumina (Pt/γ-Al2O3) for asymmetric hydrogenation for ethyl and methyl pyruvates.36,37
Major advancement in the non-metal oxide supported heterogeneous asymmetric catalyst was firstly reported by Li and coworkers11 who developed chirally modified Pt-nanoparticle supported on multi-walled carbon nanotubes. These Pt/MWNTs were found to be highly active and lead to highly enantioselective hydrogenation for various α-ketoesters. Interestingly, high enantioselectivities and TOF were obtained when Pt nanoparticles are encapsulated in nanotubes than on the surface. The high catalytic activity of Pt encapsulated CNT credited to nano-channel like behaviour that leads to greater interactive enrichment of substrate and modifier at Pt surface.11 This study motivated us to examine all other carbon based materials including MWNTs as support matrix for Pt based heterogeneous catalysis with optimal enantioselectivity at multigram scale. Herein, we report the improved synthesis of highly dispersed platinum nanoparticles loaded on functionalized carbon materials (MWNTs, CF and graphene) for hydrogenation of methyl pyruvate (representative example) using a chiral modifier. Heterogenisation is achieved by the coordination between the chiral modifier and the Pt functionalized carbon surface.38–40 We have carried out a brief and in-depth study on asymmetric hydrogenation of methyl pyruvate using Pt/C catalytic systems for achieving better insight in such transformations.
Experimental
Materials
Chloroplatinic acid (37.50% Pt basis) and MWNTs (>95% carbon) were purchased from Aldrich. Graphene, methyl pyruvate (98% assay) and cinchonidine (99% total base) was received from Alfa Aesar. Carbon fiber (Auro carbon industry), acetic acid (Fisher scientific, 99.9%), platinum on activated carbon (across, 5% Pt) and ethanol (commercial grade) were obtained commercially. High purity dry hydrogen and argon (99.999%) gas was used from local resources. BUCHI Glass Oven B-585 Kugelrohr was used to purify the reaction products.
Characterization
An X-ray diffractometer, D8 advance (Bruker, U.S.A.) using Cu Kα1 (λ = 1.54056 Å) as a radiation source was used to ascertain the quality and crystalline nature of carbon materials with tube current and voltage of 40 mA and 40 kV, respectively. Morphology of M/C was determined using scanning electron microscopy (SEM, EVO18 Ziess) with an accelerating voltage of 20 keV. The composite was pasted on silver tape for imaging. Transmission electron microscopy (TEM) was carried out in a FEI Tecnai-G2 T20. For TEM study, the catalysts were dispersed in ethanol using tip ultrasonication. The samples were prepared by placing a droplet of suspension on to a copper grid and dried in air. Diffuse reflectance spectroscopy measurement was carried on Varian Cary 4000 in the range of 200–800 nm. The electrochemical activity of Pt/MWNTs catalyst was analysed in oxygen reduction reaction by cyclic voltammetry (CH Instruments, electrochemical workstation). Enantioenriched samples were characterized by IR, UV, NMR, HPLC and polarimeter. Fourier-transform infrared spectra (FTIR) were recorded on a Vertex 70v spectrometer (Bruker) in the range of 400–4000 cm−1. UV-vis spectroscopy data were recorded on Varian Cary 4000 in the range of 200–800 nm. Nuclear magnetic resonance spectra (1H NMR) were recorded on a Bruker 500 spectrometer operating at 500 MHz in CDCl3. HPLC chromatogram was recorded on WATERS, Ireland. Chromatographic resolutions of the enantiomers were carried out on a normal phase CHIRALPAK ID (Lot No. ID00CE-PL014) type chiral column under isocratic and isothermal (at 30 °C) conditions using respective mixture of n-hexane/iso-propanol as the mobile phase. Catalytic asymmetric hydrogenation reaction was carried out in a Berghof HR-100 high pressure vessel equipped with a Teflon container. Specific rotation was measured from Rudolph polarimeter (APII/2W), purchased from USA.
Preparation of metal loaded carbon materials (M/C)
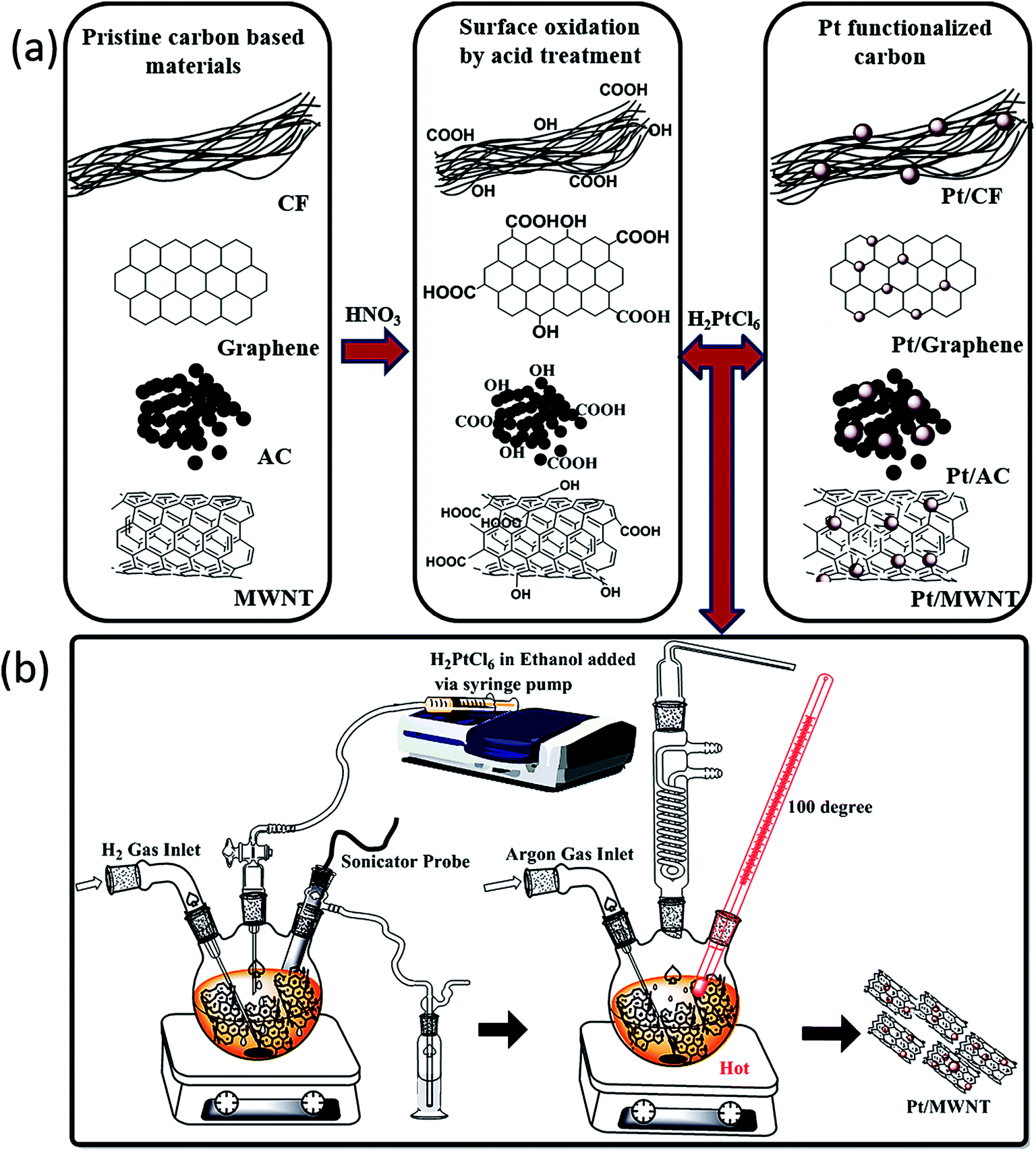
A typical preparation experiment for Pt/MWNTs has been carried out as described below and similar procedure was used for other carbon materials. First, the activation process was carried out in which MWNTs (10 g) were refluxed under constant stirring in HNO3 (500 mL, 68 wt%) at 140 °C, and extracted by centrifugation at 3000 rpm. MWNTs were washed several times with water followed by ethanol. Resultant MWNTs were dried at 80 °C for 4 h under argon.
The functionalized MWNTs were immersed in an ethanolic solution under stirring and sonication simultaneously. Platinum metal salt (5%, H2PtCl6) dissolved in degassed ethanol and added to CNT solution slowly via syringe pump under hydrogen bubbling for a period of 12 h. The solution was stirred for another 24 h at room temperature and finally heated at 100 °C for 12 h. Metal nanoparticles were tinted on carbon surface, the platinum loaded MWCNT were isolated via centrifugation, followed by washing with deionised degassed water and drying at 80 °C for 16 h under argon (Fig. 1(b)).
 |
| | Fig. 1 (a) Process for surface activation and functionalization of carbon materials (b) preparation of highly dispersed Pt nanoparticles on carbon materials. | |
Asymmetric hydrogenation of methyl pyruvate
Pt loaded catalysts were pre-treated in tubular furnace under hydrogen flow for 3 h at 300 °C, prior to reaction. For reaction, Pt/C (300 mg), chiral modifier (CD, 40 mg, 0.13 mmol), and acetic acid (15 mL, 23.7 mmol) were premixed in Teflon lined stainless steel vessel under hydrogen and after 10 minutes methyl pyruvate (0.75 mL, 8.3 mmol) was introduced. The reaction mixture was purged with hydrogen 2–3 times to remove the air and filled under the desired pressure (10–80 bar; Caution of pressurized hydrogen!) followed by stirring for 4 h. Subsequently, the reaction mixture was centrifuged to obtain the crude product. The product was purified via bulb to bulb distillation and analysed by 1H NMR and HPLC. The % yield was calculated by weighing the finally obtained purified product, using balance (Mettlar Toledo-ML204) with accuracy of 0.1 mg. The reaction was scaled up to 10 g successfully.
(R)-(+)-Methyl lactate
1H NMR (CDCl3, 500 MHz) δ: 1.41 (d, 3H, J = 7.05 Hz, CH3), 2.93 (d, 1H, J = 5 Hz OH, D2O exchangeable), 3.79 (s, 3H, CH3), 4.29 (q, 1H, J = 6.94 Hz, CH) ppm; 13C NMR (CDCl3, 125 MHz): 20.0, 52.2, 66.7, 175.7 ppm; (see ESI Fig. S12–S16†), [α]20°CD = +8.9 (c 1.5, 1, 4-dioxane);41 IR (KBr) 3346, 2975, 1710, 1379, 1266, 1084, 1052, 831 cm−1 (see ESI Fig. S9†). Optical purity was determined by HPLC (220 nm); Daicel Chiral Cell CHIRALPAK ID 0.46 cm/25 cm; hexane/iso-propanol/Na2B2O7 = 90/10/0.1; flow rate = 1.0 mL min−1; retention time: 2.9 min for (R)-(+)-methyl lactate, 3.4 min for (S)-(−)-methyl lactate (see ESI Fig. S5–S8†). UV-vis (CH3COOH); 210–250 nm (see ESI Fig. S10 and S11†).
Results and discussion
A simple process adopted for surface activation and functionalisation using HNO3 as oxidizing agent is shown schematically in Fig. 1(a). This process introduces carboxyl (–COOH), hydroxyl (–OH) and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups on to the surface of pristine carbon materials.42 These functional groups play a vital role in facilitating the binding, embedding or stacking of nanoparticles on the surface of carbon based materials.42–45 The Pt nanoparticles are formed on the activated surface from the highly diluted H2PtCl6 solution in presence of hydrogen gas as reducing agent. Functionalized carbon support and Pt metal loaded on carbon materials were characterized using XRD as shown in Fig. 2. The XRD pattern for functionalized carbon shows an intense peak at 25.90° along with other peaks at 42.70° and 53.50° corresponding to the (002), (100), (004) reflections, respectively. While the Pt loading results in three major peaks at 39.68°, 46.4° and 67.7° corresponding to diffraction from the (111), (200), and (220) planes of the face-centred cubic (FCC) lattice of Pt metal. The (100) related to carbon disappears after the loading of Pt nanoparticles. However, the XRD analysis confirms that the carbon material retain its structural planes even after functionalisation. The morphology, shape, size and distribution of nanoparticles on CF, graphene and MWNTs were characterized by SEM and TEM imaging. As seen from SEM images in Fig. 3(a)–(c), the Pt nanoparticles uniformly cover the surface of carbon support system in all the three cases. The TEM images in Fig. 3(d)–(f) show that the nanoparticles are extremely small in the size range of 2–20 nm. The nanoparticles are well dispersed in case of graphene and MWNTs as compared to carbon fibres. Graphene and MWNTs provide more reduction sites due to higher surface area. The higher magnification TEM images in Fig. 3(g)–(i) provide further information about the shape and allocation of nanoparticles on the surface of various carbon materials. The Pt nanoparticles on MWNTs are particularly interesting as the nanoparticles are seen encapsulated on the outer as well as inner surface of the MWNTs (see ESI Fig. S1a and b†). The TEM image in Fig. 3(i) shows Pt nanoparticles distinctly dispersed at few nm length scales like a single atomic system. This provides low coordination with quantum size effect and also prevents metal leaching.26 The EDX analysis shows that 4.1 ± 0.6 wt% of Pt loading takes place upon repeated synthesis of composite. (see ESI Fig. S3 and S4†). To optimize the reaction conditions, the hydrogenation of methyl pyruvate (0.8 mmol) was carried out in acetic acid (2.3 mL) using commercially available 5% Pt/AC (see ESI Fig. S2†) at room temperature by varying the pressure (Table 1). Highest product yield was obtained at 70 bar pressure. In absence of chiral modifier, racemic mixtures of methyl lactate were obtained, where in presence of CD, the enantiomeric product was separated and analysed using HPLC. Based on these observations, asymmetric catalytic hydrogenation of methyl pyruvate was studied using different catalysts under the optimized reaction conditions (Table 2). Among these catalytic systems, the Pt/MWNTs catalyst proved to be the most effective in terms of product yield and enantioselectivity i.e. >99% ee, which is highest for supported catalytic asymmetric hydrogenation of methyl pyruvate (see ESI Table S2†). The specific rotation of the product was determined using a polarimeter, which confirmed the enantioselectivity reduction to R-(+)-methyl lactate.
O) groups on to the surface of pristine carbon materials.42 These functional groups play a vital role in facilitating the binding, embedding or stacking of nanoparticles on the surface of carbon based materials.42–45 The Pt nanoparticles are formed on the activated surface from the highly diluted H2PtCl6 solution in presence of hydrogen gas as reducing agent. Functionalized carbon support and Pt metal loaded on carbon materials were characterized using XRD as shown in Fig. 2. The XRD pattern for functionalized carbon shows an intense peak at 25.90° along with other peaks at 42.70° and 53.50° corresponding to the (002), (100), (004) reflections, respectively. While the Pt loading results in three major peaks at 39.68°, 46.4° and 67.7° corresponding to diffraction from the (111), (200), and (220) planes of the face-centred cubic (FCC) lattice of Pt metal. The (100) related to carbon disappears after the loading of Pt nanoparticles. However, the XRD analysis confirms that the carbon material retain its structural planes even after functionalisation. The morphology, shape, size and distribution of nanoparticles on CF, graphene and MWNTs were characterized by SEM and TEM imaging. As seen from SEM images in Fig. 3(a)–(c), the Pt nanoparticles uniformly cover the surface of carbon support system in all the three cases. The TEM images in Fig. 3(d)–(f) show that the nanoparticles are extremely small in the size range of 2–20 nm. The nanoparticles are well dispersed in case of graphene and MWNTs as compared to carbon fibres. Graphene and MWNTs provide more reduction sites due to higher surface area. The higher magnification TEM images in Fig. 3(g)–(i) provide further information about the shape and allocation of nanoparticles on the surface of various carbon materials. The Pt nanoparticles on MWNTs are particularly interesting as the nanoparticles are seen encapsulated on the outer as well as inner surface of the MWNTs (see ESI Fig. S1a and b†). The TEM image in Fig. 3(i) shows Pt nanoparticles distinctly dispersed at few nm length scales like a single atomic system. This provides low coordination with quantum size effect and also prevents metal leaching.26 The EDX analysis shows that 4.1 ± 0.6 wt% of Pt loading takes place upon repeated synthesis of composite. (see ESI Fig. S3 and S4†). To optimize the reaction conditions, the hydrogenation of methyl pyruvate (0.8 mmol) was carried out in acetic acid (2.3 mL) using commercially available 5% Pt/AC (see ESI Fig. S2†) at room temperature by varying the pressure (Table 1). Highest product yield was obtained at 70 bar pressure. In absence of chiral modifier, racemic mixtures of methyl lactate were obtained, where in presence of CD, the enantiomeric product was separated and analysed using HPLC. Based on these observations, asymmetric catalytic hydrogenation of methyl pyruvate was studied using different catalysts under the optimized reaction conditions (Table 2). Among these catalytic systems, the Pt/MWNTs catalyst proved to be the most effective in terms of product yield and enantioselectivity i.e. >99% ee, which is highest for supported catalytic asymmetric hydrogenation of methyl pyruvate (see ESI Table S2†). The specific rotation of the product was determined using a polarimeter, which confirmed the enantioselectivity reduction to R-(+)-methyl lactate.
 |
| | Fig. 2 XRD spectra of (a) Pt loaded carbon materials (b) functionalized carbon materials. | |
 |
| | Fig. 3 SEM and TEM image of Pt nanoparticles loaded on carbon materials surface (a, d, g) Pt/CF (b, e, h) Pt/graphene (c, f, i) Pt/MWNT. | |
Table 1 Optimization of reaction conditions using commercial Pt loaded activated carbon (Pt/AC) for hydrogenation of methyl pyruvatea

|
| S. No. |
Pressure (bar) |
Yielda (%) |
| Reaction was carried out using 5% Pt/AC (4 mg) methyl pyruvate (0.8 mmol) and acetic acid (2.3 mL) for 4 h at room temperature. Racemic mixtures of the products were obtained. |
| 1 |
10 |
60 |
| 2 |
20 |
80 |
| 3 |
30 |
90 |
| 4 |
40 |
92 |
| 5 |
50 |
94 |
| 6 |
60 |
96 |
| 7 |
70 |
97 |
| 8 |
80 |
90 |
Table 2 Asymmetric hydrogenation of methyl pyruvate using various catalytic systemsa
| S. No. |
Catalyst |
Yield (%) |
eeb (%) |
| Pt loading for all catalyst is 5 wt%. Reaction condition: methyl pyruvate/acetic acid/CD/hydrogen pressure/time/temperature: 0.75 mL/15 mL/40 mg/70 bar/4 h/RT. Determined by HPLC analysis using a CHIRALPAK ID chiral column. The optical yield was expressed as the enantiomeric excess (ee) of R-(+) enantiomer% ee = ([R] − [S])/([R] + [S]) × 100 (see ESI Fig. S5–S8). |
| 1 |
Pt/AC |
97 |
31 |
| 2 |
Pt/graphene |
87 |
91 |
| 3 |
Pt/carbon fibre |
75 |
89 |
| 4 |
Pt/MWNT |
99 |
>99 |
Substrate–modifier–catalyst interaction
While exploring the possible mechanism of the asymmetric hydrogenation (Fig. 4), it is generally believed that CD, substrate and hydrogen are absorbed concertedly on the Pt surface which provokes the enantioselectivity.46–51 The in situ 1H NMR was recorded in toluene-d8 as reaction progresses to understand the substrate–modifier interactions (see ESI Fig. S17†). We have observed significant changes in chemical shift for H2, H6, H9, H5′ and H8′ protons when CD and methyl pyruvate are combined. H2 and H6 proton goes to downfield due to hydrogen bonding between the adsorbed CD modifier quinuclidine N and O atom of the methyl pyruvate (see ESI Fig. S17d†). Proton H9 also showed downfield shift, see Fig. 5(a) and (b). This chemical shift of proton (H9) shows interaction between OH group of CD and O atom of methyl pyruvate. Thus, overall NMR study suggested that besides aliphatic part of CD, the aromatic ring (quinoline) is also involved in the formation of the methyl pyruvate–CD interaction via π-electrons.47,48 The NMR studies also support π–π communication between conjugated double bond of aromatic rings of CD (Fig. 5). Fig. 5(c) and (d) shows chemical shift of H5′ and H8′ proton where it was observed that H8′ proton goes to downfield and H5′ proton goes to upfield. The former interaction (quinuclidine N and O atom of methyl pyruvate) is stronger than the π–π interaction mentioned above, however, the π–π interaction might be sufficient to alter the chemical shift recorded in NMR with time (see ESI Table S1†). Both type of interactions help to control the mode of adsorption of the substrate and chiral induction. Thus, cinchonidine behaves like a bidentated ligand and form substrate–modifier complex.
 |
| | Fig. 4 Intermediate structure of asymmetric hydrogenation reaction of methyl pyruvate on catalyst surface. | |
 |
| | Fig. 5 NMR studies of (a) NMR spectra for H9 proton and corresponding (b) chemical shift plotted with respect to reaction time. (c) NMR spectra for H5′ & H8′ proton and corresponding (d) chemical shift versus reaction time. | |
UV-DRS spectra of CD, Pt/CD/MWNTs, and Pt/MWNTs are shown in Fig. 6. As observed, CD produces a steep absorption edge at about 350 nm with poor absorption. Whereas, Pt/MWNTs shows a sharp absorption edge at 375 nm with enhanced absorption in visible region at 608 nm and 669 nm due to localized surface plasmon resonance (LSPR) associated with platinum nanoparticles. In the presence of CD, the UV-DRS spectrum of Pt/MWNTs/CD shows reduced absorption due to the bonding of Pt nanoparticles with CD. Pt/MWNTs is observed to exhibit high absorption among all three Pt/C composites (see ESI Fig. S18†).
 |
| | Fig. 6 DRS spectrum of (a) CD (b) Pt/CD/MWNT (c) Pt/MWNT. | |
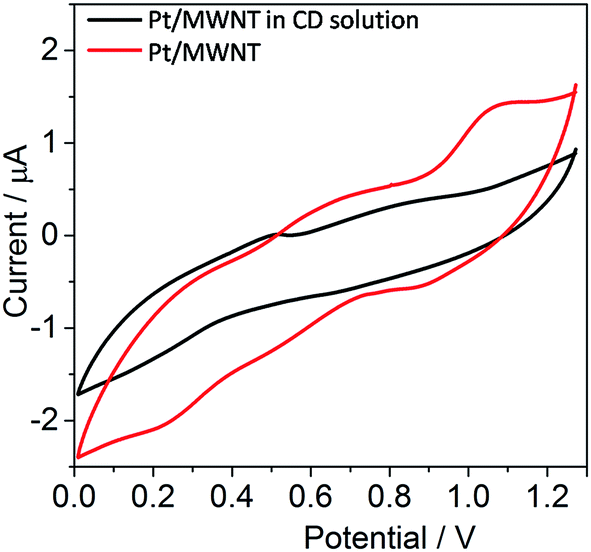
Adsorption of CD on Pt surface has been investigated by cyclic voltammetry (Fig. 7). The CV measurement of the CD (10−4 mol L−1) in H2SO4 exhibited decrease in current as compared to Pt/MWNTs measured in pure 0.5 mol L−1 H2SO4. This shows that CD adsorption take place on the Pt surface and inhibits the participation of Pt metal in redox process.52,53 Current observations manifested that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of substrate/modifier absorb on Pt surface to make asymmetric hydrogenation reaction successful.49,50 Various interactions brought the stereogenic centre of CD close enough to the substrate to achieve high enantioselectivity of 99%. A pictorial hypothesis of intermediate is shown in Fig. 4, where a favourable transition state with both stereogenic centres of CD is being proposed.
:1 complex of substrate/modifier absorb on Pt surface to make asymmetric hydrogenation reaction successful.49,50 Various interactions brought the stereogenic centre of CD close enough to the substrate to achieve high enantioselectivity of 99%. A pictorial hypothesis of intermediate is shown in Fig. 4, where a favourable transition state with both stereogenic centres of CD is being proposed.
 |
| | Fig. 7 Cyclic voltammetry of Pt/MWNT and CD adsorb Pt/MWNT in H2SO4 solution. | |
Recyclability of catalyst
Heterogeneous catalysts are more stable, which signify its importance in organic synthesis.54 In present case, the stability of Pt/MWNTs as catalyst was tested by recycling process, where the catalyst was separated out by centrifugation and washed with acetic acid after each reaction cycle. Fresh reactant and modifier were added to the regenerated catalyst and steps were repeated for 10 consecutive cycles. The regenerated catalyst provided low enantioselectivity in absence of modifier. A relatively constant conversion and enantioselectivity was obtained in all cycles (Table 3). The HRTEM images of recycled catalyst (after ten cycles) revealed individuality of catalytic reaction centre that secured high enantioselectivity (See ESI Fig S19†).
Table 3 Recyclability of catalyst (Pt/MWNT)a
| Entry (cycle) |
Yield (%) |
eeb (%) |
| Pt loading for all catalysts is 5 wt%. Reaction condition: methyl pyruvate/acetic acid/CD/hydrogen pressure/time/temperature: 0.75 mL/15 mL/40 mg/70 bar/4 h/RT. Determined by HPLC analysis using a CHIRALPAK ID chiral column. |
| 1 |
98.5 |
>99 |
| 2 |
98.3 |
99 |
| 3 |
96.4 |
>97 |
| 4 |
95.9 |
97 |
| 5 |
94.4 |
97 |
| 6 |
94.3 |
96 |
| 7 |
94.1 |
96 |
| 8 |
93.9 |
96 |
| 9 |
93.3 |
94 |
| 10 |
93.0 |
94 |
Conclusion
In conclusion, asymmetric heterogeneous hydrogenation of methyl pyruvate is a representative example of α-ketoester has been studied using Pt/C as catalytic system. The Pt/MWNTs was found to be most efficient and provided highest enantioselectivity. The supremacy of Pt/MWNTs is attributed to the high absorption of CD/substrate on atomically dispersed Pt nanoparticles loaded on nanochannels of MWNTs that maximized metal utilization. While NMR studies elucidated 1:1 substrate modifier complex, cyclic voltammetry suggested irreversible adsorption of CD on Pt surface. The catalyst is reusable without any significant loss of activity even after ten cycles. Furthermore, significant attempts have being made to understand the mechanism and kinetics of such reactions using NMR. Current study provides an opportunity to develop large scale industrially viable asymmetric hydrogenation catalysts.
Acknowledgements
We thank Indian Institute of Technology Jodhpur, India for a studentship (to PS), and DST-RFBR (Indo-Russian bilateral project INT/RFBR/P-134) for financial support and Dr Ritu Gupta (IIT Jodhpur) for useful discussions. Dedicated to Prof. Ashoka G. Samuelson for his teaching and guidance on the occasion of his 60th birthday.
References
- M. Titirici, R. J. White, N. Brun and V. L. Budarin, Chem. Soc. Rev., 2015, 44, 250 RSC.
- D. M. Sun, C. Liu, W. C. Ren and H. M. Cheng, Small, 2013, 9, 1188 CrossRef CAS PubMed.
- D. Janas and K. K. Koziol, Nanoscale, 2014, 6, 3037 RSC.
- G. Xiong, C. Meng, R. G. Reifenberger, P. P. Irazoqui and T. S. Fisher, Energy Technol., 2014, 2, 897–905 CrossRef CAS.
- M. Fraga, M. Mendes and E. Jordao, J. Mol. Catal. A: Chem., 2002, 179, 243–251 CrossRef CAS.
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed.
- H. Guoa, Y. Zhua, W. Lia, H. Zhenga, K. Wua, K. Dinga, B. Ruana, A. Hagfeldtb, T. Mac and M. Wua, Electrochim. Acta, 2015, 176, 997–1000 CrossRef.
- W. Chen, J. Ji, X. Feng, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, J. Am. Chem. Soc., 2014, 136, 16736–16739 CrossRef CAS PubMed.
- S. H. Hsieha, M. C. Hsub, W. L. Liua and W. J. Chen, Appl. Surf. Sci., 2013, 277, 223–230 CrossRef.
- R. S. Oosthuizen and V. O. Nyamori, Platinum Met. Rev., 2011, 55, 154–169 CrossRef CAS.
- Z. Chen, Z. Guan, M. Li, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2011, 50, 4913–4917 CrossRef CAS PubMed.
- J. Pritchard, A. G. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- B. Minder, T. Mallat, K. Pickel, K. Steiner and A. Baiker, Catal. Lett., 1995, 34, 1–9 CrossRef CAS.
- M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 CrossRef CAS PubMed.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- G. Szollosi, B. Hermán, F. Fülöp and M. Bartók, J. Catal., 2010, 276, 259–267 CrossRef CAS.
- M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed.
- Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Handbook of Asymmetric Heterogeneous Catalysis, ed. K. Ding and Y. Uozumi, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. Hong, I. Lee and F. Zaera, Catal. Sci. Technol., 2015, 5, 680–689 CAS.
- A. Wilhelmus, H. Vermeer, A. Fulford, P. Johnston and P. B. Wellset, J. Chem. Soc., Chem. Commun., 1993, 1053–1054 Search PubMed.
- H. U. Blaser and H. P. Jalett, Stud. Surf. Sci. Catal., 1993, 78, 139–146 CrossRef CAS.
- H. U. Blaser, H. P. Jalett, M. Muller and M. Studer, Catal. Today, 1997, 37, 441–463 CrossRef CAS.
- H. U. Blaser, H. P. Jalett, W. Lottenbach and M. Studer, J. Am. Chem. Soc., 2000, 122, 12675–12682 CrossRef CAS.
- J. Kohler and J. S. Bradley, Catal. Lett., 1997, 45, 203–208 CrossRef CAS.
- Y. Orito, S. Imai and S. Niwa, J. Chem. Soc. Jpn., 1979, 1118 CrossRef CAS.
- Y. Orito, S. Imai, S. Niwa and N. G. Hung, J. Synth. Org. Chem., 1979, 37, 173 CrossRef CAS.
- E. Talas, J. L. Margitfalvi and O. Egyed, J. Catal., 2009, 266, 191–198 CrossRef CAS.
- P. B. Wells and R. P. K. Wells, in Chiral Catalyst Immobilization and Recycling, ed. D. E. de Vos, I. F. J. Vankelecom and P. A. Jacobs, Wiley–VCH, Weinheim, 2000, p. 123 Search PubMed.
- D. Y. Murzin, P. Maki-Arvela, E. Toukoniitty and T. Salmi, Catal. Rev.: Sci. Eng., 2005, 47, 175 CAS.
- M. Bartók, Curr. Org. Chem., 2006, 10, 1533 CrossRef.
- J. L. Margitfalvi and E. Talas, in Asymmetric hydrogenation of activated ketones, ed. J. J. Spivey and K. M. Dooley, RSC, Cambridge, 2010, vol. 22, pp. 144–278 Search PubMed.
- M. Studer, H. U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS and references cited therein.
- Y. Orito, S. Imai and S. Niwa, Collected papers of the 43rd Catalysis Forum, Japan, 1978, p. 30 Search PubMed.
- S. Bhaduri, G. K. Lahiri, P. Munshi and D. Mukesh, Catal. Lett., 2000, 65, 61–66 CrossRef CAS.
- X. Li, X. You, P. Ying, J. Xiao and C. Li, Top. Catal., 2003, 25, 63–70 CrossRef CAS.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC.
- J. M. Fraile, J. I. Garcia, J. A. Mayoral and T. Tarnai, Tetrahedron: Asymmetry, 1998, 9, 3997–4008 CrossRef CAS.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- R. K. Sharma and A. G. Samuelson, J. Chem. Sci., 2006, 118, 569–573 CrossRef CAS.
- C. J. Abraham, D. H. Paull, T. Bekele, M. T. Scerba, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2008, 130, 17085–17094 CrossRef CAS PubMed.
- M. von Arx, N. Dummer, D. J. Willock, S. H. Taylor, R. P. Wells, P. B. Wells and G. J. Hutchings, Chem. Commun., 2003, 48, 1926–1927 RSC.
- B. Satishkumar, E. M. Vogl, A. Govindaraj and C. Rao, J. Phys. D: Appl. Phys., 1996, 29, 3173–3176 CrossRef CAS.
- S. Tsang, Y. Chen, P. Harris and M. Green, Nature, 1994, 372, 159–162 CrossRef CAS.
- E. Dujardin, T. Ebbesen, H. Hiura and K. Tanigaki, Science, 1994, 265, 1850–1852 CAS.
- M. Garland and H. U. Blaser, J. Am. Chem. Soc., 1990, 112, 7048–7050 CrossRef CAS.
- T. A. Martinek, T. Varga, F. Fulop and M. Bartok, J. Catal., 2007, 246, 266–276 CrossRef CAS.
- A. Baiker, J. Mol. Catal. A: Chem., 2000, 163, 205 CrossRef CAS.
- O. Schwalm, B. Minder, J. Weber and A. Baiker, Catal. Lett., 1994, 23, 271 CrossRef CAS.
- K. E. Simons, P. A. Meheux, S. P. Griffiths, I. M. Sutherland, P. Johnston, P. B. Wells, A. F. Carley, M. K. Rajumon, M. W. Roberts and A. Ibbotson, Recl. Trav. Chim. Pays-Bas, 1994, 113, 465 CrossRef CAS.
- D. Ferri, T. Buergi and A. Baiker, J. Catal., 2002, 210, 160–170 CrossRef CAS.
- I. Bakos, S. Szabo, M. Bartok and E. Kalman, J. Electroanal. Chem., 2002, 532, 113 CrossRef CAS.
- S. Lavoie, M. A. Laliberte, I. Temprano and P. H. McBreen, J. Am. Chem. Soc., 2006, 128, 7588–7593 CrossRef CAS PubMed.
- V. Morawsky, U. Prusse, L. Witte and K. D. Vorlop, Catal. Commun., 2000, 1, 15–20 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21790c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.