
Spirochromone-chalcone conjugates as antitubercular agents: synthesis, bio evaluation and molecular modeling studies†
M. Mujahida,
P. Yogeeswarib,
D. Sriramb,
U. M. V. Basavanagc,
Erik Díaz-Cervantesc,
Luis Córdoba-Bahenac,
Juvencio Roblesc,
R. G. Gonnadea,
M. Karthikeyana,
Renu Vyasa and
M. Muthukrishnan*a
aCSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune, Maharashtra 411008, India. E-mail: m.muthukrishnan@ncl.res.in; Fax: +91-20-25902629; Tel: +91-20-25902284
bMedicinal Chemistry & Antimycobacterial Research Laboratory, Pharmacy Group, Birla Institute of Technology & Science – Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad 500 078, India
cDepartamento de Química y Departamento de Farmacia, Universidad de Guanajuato, Noria Alta S/N, Col. Noria Alta, C. P. 36050, Guanajuato, Gto, Mexico
First published on 2nd December 2015
Abstract
A new series of spirochromone annulated chalcone conjugates were synthesized and evaluated for their antitubercular activity against Mycobacterium tuberculosis H37Rv strain. These compounds were subjected to molecular modeling studies using docking and chemoinformatics based approaches. The docking simulations were performed against a range of known receptors for chalcone derived compounds to reveal MTB phosphotyrosine phosphatase B [MtbPtpB] protein as the most probable target based on the high binding affinity scores. Five compounds exhibit significant inhibition, showing minimum inhibitory concentration values i.e. MIC values ranging from 3.13–12.5 μg mL−1. Further analysis of the synthesized compounds with known and in-house developed chemoinformatics tools unequivocally established their potential as anti-tubercular compounds. QSAR modeling revealed a quantitative relationship between biological activities and frontier molecular orbital energies of synthesized compounds. The predictive model can be employed further for virtual screening of new compounds in this series.
Introduction
Tuberculosis is a devastating bacterial disease caused by Mycobacterium tuberculosis (Mtb). It poses a major public health risk due to the long duration of treatment (6–9 months), co-infection with HIV and the emergence of multidrug resistant strains etc. According to a WHO report, in 2013 alone, nearly 9 million people contracted tuberculosis and 1.5 million died from complications of the disease.1 Despite enormous efforts to develop anti-mycobacterial drug molecules, tuberculosis still constitutes the leading killer disease. Therefore, the discovery and development of new types of anti TB agents acting on novel drug targets is urgently needed.It is a well known fact that biologically active natural products are recognized as evolutionarily selected and biologically prevalidated starting point for any successful drug discovery program. Therefore, the synthesis of natural products inspired compound collections and their biological evaluation is a highly promising strategy for the identification of unique biologically relevant compound classes, especially in the studies aimed at finding a new class of anti TB agents.2 Flavonoids are the most explored class of natural products as they are widely distributed in various plants and foods such as fruits & vegetables.3 Chalcones are an important sub class of flavonoids that consist of open chain flavonoids in which the two aromatic rings are fused by a three-carbon α,β-unsaturated carbonyl system.4 They have been reported to possess plethora of medicinal applications which include anti-inflammatory, antileukemic, antiulcerogenic, antimalarial, antileishmaniasis, and anticancer activities.5 Interestingly, many natural and natural product inspired chalcone analogues exhibit significant antimycobacterial activity (Fig. 1).6 Recently, several research groups successfully demonstrated their mode of action by identifying their molecular targets.7
 | ||
| Fig. 1 Natural/NP inspired chalcones with potent activity against Mtb. | ||
A good safety profile, possibility of oral administration,8 and easy synthesis are the major factors contributing to the growing interest in exploring the pharmacological activities of chalcones. In this context, and in view of our long standing interest in the chemistry of the privileged chromone motif,9 we recently reported various spirochromone derivatives possessing 1,2,3-triazole and amino alcohol moieties that can serve as lead molecules for developing antitubercular agents.10 These results encouraged us to further explore the spirochromone motif as an active pharmacophore for further diversification to exploit its anti TB potential. Herein, we describe the synthesis of novel spirochromone annulated chalcones, their in silico studies and in vitro screening results against Mycobacterium tuberculosis H37Rv (Fig. 2). To the best of our knowledge, no reports on the synthesis, in silico study and antimycobacterial activities of these spirochromone annulated chalcone have been reported.
 | ||
| Fig. 2 Design of spirochromone annulated chalcone conjugates. | ||
Results and discussion
Synthesis
A three step synthetic strategy was followed for the preparation of spirochromone annulated chalcones as outlined in Scheme 1. As shown, the readily available resorcinol 1 on heating with acetic anhydride in the presence of anhydrous zinc chloride provided 4,6-diacetyl resorcinol 2 in 75% yield.11 The diacetyl compound 2 on Kabbe condensation with cyclohexanone or N-Boc piperidone in the presence of pyrrolidine as a base in refluxing toluene using Dean–Stark apparatus provided spirochromone derivatives 3a and 3b in 62% and 65% yields. Lastly, spirochromone derivatives 3a,b on treatment with various aryl aldehydes using Claisen–Schmidt condensation reaction produced spirochromone annulated chalcones 4a–g & 5a–h in moderate to good yields. The structure of all the new products 4 & 5 (15 compounds) was confirmed by 1H NMR, 13C NMR and mass spectral analysis. For an example, in the 1H NMR spectra (compound 4f as a representative example), a signal corresponding to the C-3 protons of chromone skeleton was observed as a singlet at δ 2.73 ppm and the corresponding 13C resonance signal was observed at δ 47.8 ppm and the spirocarbon was discernible at δ 81.4 ppm. Further, the appearance of a sharp singlet (1H) observed at δ 13.5 ppm in the PMR, suggested the presence of one chelated hydroxyl group. In addition, the appearance of two sets of doublets at δ 7.61 & 7.87, 1H each with coupling constant 15.4 Hz were ascribable to the olefinic protons of the chalcone moiety. Conclusive evidence for its structure was derived from a single crystal X-ray analysis (Fig. 3). | ||
| Scheme 1 Reagents and conditions: (i) Ac2O, ZnCl2, 140 °C, 30 min, 75%; (ii) cyclohexanone/N-Boc-piperidone, pyrrolidine, toluene, reflux, 12 h; (iii) aromatic aldehyde, 50% aq. KOH, MeOH. | ||
 | ||
| Fig. 3 ORTEP diagram of the compound 4f. | ||
Anti-mycobacterial evaluation
All the new spirochromone annulated chalcone conjugates were screened for their in vitro antimycobacterial activity against M. tuberculosis H37Rv (ATCC27294) using an agar dilution method. The minimum inhibitory concentration (MIC; μg mL−1) was determined for each compound. The MIC is defined as the minimum concentration of compound required to completely inhibit the bacterial growth. Rifampicin and ethambutol were used as reference compounds. The MIC values of the synthesized compounds along with the standard drugs for comparison are reported in Table 1.| Entry | Compound | MIC (μg mL−1) | Entry | Compound | MIC (μg mL−1) |
|---|---|---|---|---|---|
| 1 |  |
12.5 | 10 |  |
3.13 |
| 2 | 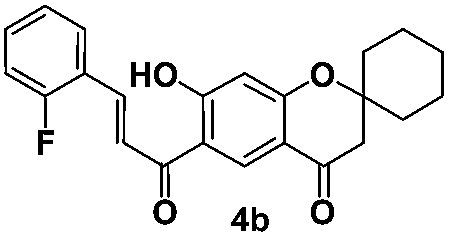 |
12.5 | 11 |  |
12.5 |
| 3 |  |
25 | 12 | 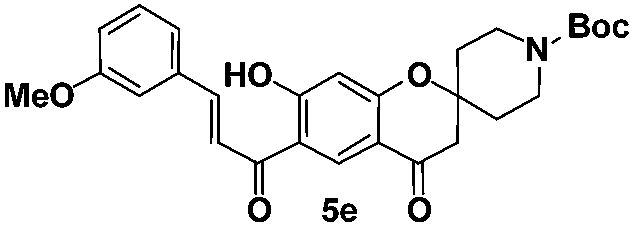 |
25 |
| 4 |  |
>25 | 13 | 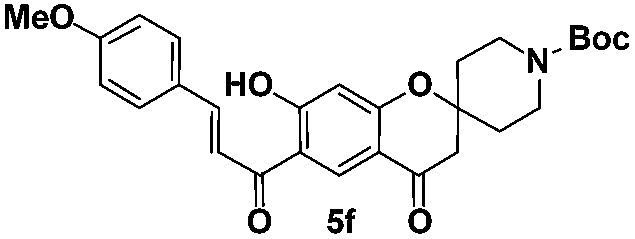 |
25 |
| 5 | 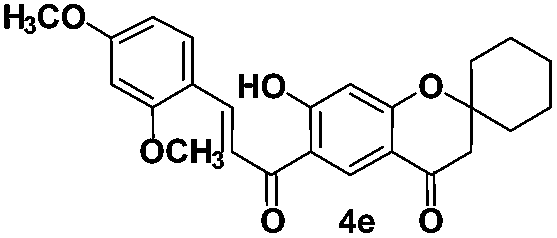 |
>25 | 14 |  |
25 |
| 6 |  |
>25 | 15 |  |
>25 |
| 7 |  |
>25 | 16 | Rifampicin | 0.2 |
| 8 |  |
>25 | 17 | Ethambutol | 1.56 |
| 9 | 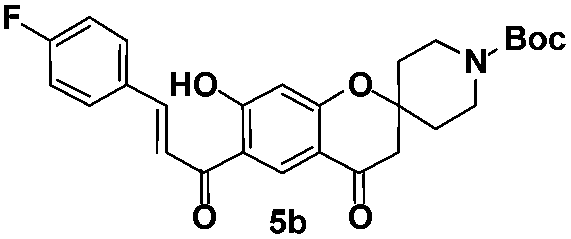 |
6.25 |
Among the 15 spirochromone annulated chalcone conjugates tested, five compounds 4a, 4b, 5b, 5c, and 5d were shown to be active with MIC values in the range of 3.13–12.5 μg mL−1. The compound 5c is shown to be highly active among all the compounds tested with a MIC value of 3.13 μg mL−1. It has a core spirochromone moiety hitherto unreported. The preliminary SAR of the spirochromone annulated conjugates reveals that compounds possessing piperidinyl group at 2nd position of the chromone ring favors better activity than the cycloalkyl group. In addition, the activity is being influenced by functional groups present in the chalcone moiety as well. Halogen substitution at aromatic ring, especially fluoro substitution enhances the activity (5b, 5c). It is also observed that the presence of methoxy substitution at aromatic ring doesn't seem to influence the activity.
Computational studies
Methodology
Molecular docking
The molecular docking was performed and analyzed using two state of art docking algorithms AutoDock Vina13 and MOE.14 Lamarckian genetic algorithm method implemented in the program suite was employed to identify appropriate binding modes and conformation of the ligand molecules. The grid map was centered at the active site of the protein by Autogrid. The Lamarckian genetic algorithm, the pseudo-Solis and Wets methods were applied for minimization using default parameters. Binding energy, intermolecular amino acids and their distance were recorded in each ligand bound conformations. In the MOE docking simulations, the ligands were allowed flexibility, London dG scoring function was employed and top 30 conformations were retained.Molecular docking analysis
With an objective to explore the potential putative targets of the synthesized compounds among the validated Mycobacterium receptors, we performed a docking analysis with known biological targets reported so far for the chalcone scaffold derived anti-tubercular compounds. In general, the validated Mtb targets for this class of compounds are phosphotyrosine phosphatases (MtbPtp A and B; PDB ID: 1U2P, 2OZ5), dihyrdofolate reductase (DHFR), PDB ID: 1DG5) and enol acyl carrier reductases (PDB ID: 2NSD) (Table 2).15 Accordingly, automated docking was used to assess the binding modes and conformation of the compounds synthesized in this study. Among the 15 spirochromone-chalcone structures, compounds 4a, 4b, 5b, 5c and 5d were considered for docking simulations since they exhibited higher anti-tubercular activity, as ascertained from their MIC data (Table 1). The four spirochromone chalcones were cross docked against all potential protein targets to reveal better binding affinities with 2OZ5, a MTB phosphotyrosine phosphatase B [MtbPtpB] protein (Table 3). PtpB and PtpA are protein tyrosine phosphatases, a class of regulatory enzymes that are secreted by Mtb into the cytosol of infected macrophages. MtbPtpB catalyzes the dephosphorylation of serine/threonine or tyrosine and also has phosphoinositide phosphatase activity whereas PtpA specifically dephosphorylates the tyrosine residues. PtpA inhibits macrophages microbial activity by entering into host macrophages, dephosphorylates the cytoplasmic protein VPS33B and inhibits the phagosome maturation.16 MtbPtpB blocks the signal regulated kinase and p-38 mediated by IL-6 thereby promoting mycobacterial survival in the host.| PDB target | Compound 4a | Compound 4b | Compound 5b | Compound 5c | Compound 5d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MOE | Autodock Vina | MOE | Autodock Vina | MOE | Autodock Vina | MOE | Autodock Vina | MOE | Autodock Vina | |
| 1U2P | −10.00 | −7.10 | −9.65 | −6.80 | −9.98 | −6.90 | −10.15 | −7.60 | −11.25 | −8.00 |
| 2OZ5 | −12.41 | −8.16 | −12.37 | −8.30 | −13.90 | −8.90 | −13.96 | −9.00 | −11.36 | −8.30 |
| 2NSD | −12.20 | −8.10 | −11.84 | −7.80 | −12.95 | −8.30 | −12.37 | −7.10 | −12.51 | −6.80 |
| 1DG5 | −12.79 | −8.40 | −12.47 | −8.28 | −12.41 | −8.00 | −11.80 | −8.00 | −10.40 | −7.90 |
| Entry | Compound ID | Intermolecular distance (Å) | Amino acids involved in intermolecular interactions | Ligand atom | Receptor atom | Binding energy (kcal mol−1) |
|---|---|---|---|---|---|---|
| 1 |  |
1.40 | Tyr125 | H2572 | OH1069 | −9.2 |
| 1.01 | Arg59 | O2560 | NH543 | |||
| 3.48 | Arg166 | C2512 | NE1475 | |||
| 3.07 | Ser57 | C2528 | OG522 | |||
| 2.62 | Glu129 | O2558 | CG1104 | |||
| 2 |  |
2.96 | Ser57 | O2536 | OG522 | −8.2 |
| 2.96 | Ser57 | O2536 | OG522 | |||
| 2.89 | His94 | O2534 | ND847 | |||
| 2.44 | Arg166 | O2541 | NH1481 | |||
| 3 |  |
2.31 | His94 | H2537 | O843 | −8.3 |
| 2.76 | Tyr125 | O2541 | O1069 | |||
| 4 |  |
3.53 | Arg166 | O2512 | NH1481 | −8.9 |
| 5 |  |
2.52 | Ser57 | O18 | OG585 | −9.0 |
| 2.52 | Ser57 | O18 | OE621 | |||
| 2.23 | Glu60 | H43 | OG585 | |||
| 6 |  |
2.78 | Ser57 | O2543 | OG522 | −8.3 |
| 2.55 | Lys164 | O2527 | NZ1454 | |||
| 7 | 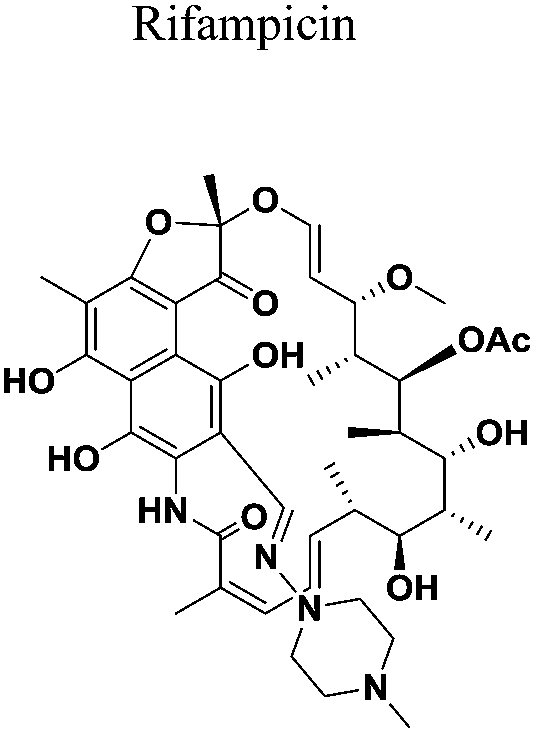 |
1.69 | Glu60 | H2620 | Oe557 | −7.0 |
| 3.53 | Phe80 | H2627 | O738 | |||
| 1.40 | Tyr125 | O2519 | OH1069 | |||
| 2.48 | Tyr125 | O2522 | OH10c69 | |||
| 2.76 | Arg166 | O2520 | NH1478 | |||
| 2.94 | Arg166 | O2521 | NH1478 | |||
| 8 |  |
3.07 | Arg166 | O2512 | N1422 | −7.8 |
| 2.41 | Phe161 | O2512 | NH1481 |
PtpA specifically PtpA and PtpB proteins share a structural homology and the active site of the phosphatase enzymes lies in the P-loop region. However, there are certain differences in the P-loop region and some sequential and positional variations occur in the variable loop. These subtle distinctions in the active sites may account for the high selectivity of the five compounds to MtbPtpB (PDB ID: 2OZ5) than MtbPtpA (PDB ID: 1U2P). Moreover the active site volume computed (CASTp server) showed a greater volume of the binding pocket of PtpB (1509.9 A3) relative to PtpA (108.7 A3) enzyme. The present compounds possess volumes ranging from 300–400 A3 which enables a better fit in the binding pocket of B compared to A. Compound 5c performed better in the docking run when compared with other synthesized compounds with binding scores ranging from −7.6 to −8.0 kcal mol−1 against the resolved crystal structure of 2OZ5. In another docking run, the synthesized molecule 5c chosen for computational studies showed comparable performance with the native ligand of receptor (entry 1, Table 3) and slightly better performance than the known MTB drug molecules rifampicin and ethambutol in terms of binding efficiencies (entries 5 and 6, Table 3). It is to be noted here that the compound 5c also possesses the best inhibitory activity in the Mtb assay. Further, the top docked conformation of this compound depicted a greater alignment with the native ligand pose (Fig. 6B). Hence it is likely to act as good binder for the phosphatase receptor in mycobacterium.
The key intermolecular interactions between selected compounds and known drugs against the phosphatase B enzyme are depicted in Fig. 4. Tyr125, Arg56, Arg166, Ser57 and Glu129 amino acids in the active site interacted with native ligand with many residues depicting a high ligand exposure in the interaction map.17 Tyr125 acts as hydrogen donor whereas Arg66 acts as hydrogen acceptor and forms a bond with the O group of the ligand. Carbonyl and hydroxyl group based interactions were common in most of the predicted binding poses of compounds in the active site pocket. Specifically, Ser57 of compound 4a, Glu60 of compound 5c, Phe80 and Arg166 of rifampicin and Arg166 and Phe161 of ethambutol made contacts with the hydroxyl group. Thr125 residue in the receptor pocket interacted with the two carbonyl groups present in the rifampicin structure (entry f, Fig. 4). Apart from hydrogen bonding, other stabilizing intermolecular interactions such as π-cation interaction were also observed.
 | ||
| Fig. 4 2D protein ligand interaction maps of compounds 4a, 4b, 5b–5d, native molecule and known tuberculosis drugs ethambutol and rifampicin. | ||
Arginine residue was mainly involved in π-cation interaction in the pocket region which helped to stabilize the protein ligand complex formation. Arg166 and Tyr125 were found to interact with the oxygen atom of the pyranone ring system in compounds 4a and 4b. Compound 5b showed a slightly lower docking energy than 5c (entry 4, Table 3). Interestingly, both possess similar structures, the only difference being the placement of the fluorine atom in the ring. Perhaps the position of the fluorine atom in the former probably hinders effective binding with the pocket residue forcing the molecule to adopt a conformation that allows only a single π-cation interaction with arginine 166 (entry f, Fig. 4). It is known from the X-ray crystal structure that the pocket region of 2OZ5 is present in a P loop region flanked by alpha helix chains α7, α8 and α3A displayed in Fig. 6A. This comparable lid of α7–α8 hairpin in located on one side of the entrance to the active site and helix α3A on the hydrophobic surface of pocket region and protects the catalytic Cys160 amino acid present within the P-loop PtpB resists oxidative inactivation better than the PtpA enzyme that does not possess this comparable lid.18 As observed in Fig. 5a, the more potent compound 5c is placed nearer to the important catalytic residue Cys160 in the binding pocket whereas the aromatic ring of 5b lies in a different plane (Fig. 5b).
 | ||
| Fig. 5 Stereo view depicting the binding mode of compounds 5b and 5c that showed high in vitro anti-mycobacterial activity. (a) depicts compound 5c in magenta, the pocket residues are shown in cyan. (b) depicts compound 5b in green along with the neighbouring residues (cyan). | ||
Compounds 4a, 5c, 5d, rifampicin and ethambutol interacted with Arg166 and Ser57 residues in the pocket region of phosphotyrosine phosphatase B protein. After post filtering process, the top docked protein conformation for compound 5c was chosen. Fig. 6B highlights the top docked pose of compound 5c aligned to the native ligand pose. Structure alignment of conformations was performed using Pymol v1.7.6. Similarity in alignment between the compounds suggests that the top docked conformation of 5c is closer to the native bioactive conformation. Fig. 6C shows the orientations of the compounds 5c and native ligand in the binding pocket of the receptor.
 | ||
| Fig. 6 Superposed docking poses for (A) pocket region of 2OZ5 with loops (green) flanked by alpha helices (red) and beta sheets (yellow). (B) Structure alignment of docked conformation of Compound 5c (magenta) with native ligand conformation (yellow) in the crystal structure. (C) Compound 5c and native ligand in the binding pocket of receptor. The receptor surface shown is coloured according to the electrostatic potential. | ||
Chemoinformatics analysis
A druggability check was performed for all the 15 synthesized compounds (Table 4). Lipinski rule of 5 predictions were performed using the Screening Assistant 2 tool.19 Most compounds displayed no violation of the standard rule of 5 indicating that they possess good drug like properties.20 The notion that these compounds could be further developed as anti-tubercular compounds was further affirmed by ADME properties prediction using PreADMET software.21 The compounds clearly lie in the acceptable range of BBB model suggesting that the compounds can penetrate the blood brain barrier. It is well established fact that for a compound to be accepted in an oral dosage form, Caco2 cell permeability values should be above 25 nms22 and the Human Intestinal Absorption (HIA) quantities should lie in the 50–100% range.23 The selected compound 5c, prioritized in this study fulfilled the above criteria. TPSA (topological polar surface area) results indicated satisfactory values for all the 15 compounds. LAZAR (lazy structure activity relationships) software detects mutagenic and/or carcinogenic properties based on the similarities in functional group with mutagenic and/or carcinogenic compounds present in the Lazar database.24 Based on this software, all the compounds were predicted to be non-carcinogenic and non-mutagenic wherein the confidence value greater than 0.025 suggested the model to give highly reliable predictions. Thus most synthesized compounds predicted favorable ADME predictions. To further corroborate our studies, an in-house developed integrated suite of programs – ChemScreener was employed. The program rapidly annotates huge chemical libraries with more than thousand pharmacophoric (P), toxicophoric (T) and chemophoric (C) attributes to prioritize compounds in a virtual screening run.25 The significance of the presence of pharmacophore and toxicophore features in a molecule is commonly known in the medicinal chemistry literature, the term ‘chemophore’ refers to groups that are too reactive or inert. A judicious selection of these features in a molecule will impact its further development as a drug. The ChemScreener results for the compounds 5b, 5c and 5d yielded a PTC score of 56, 33 and 25 indicating that they possess an optimum combination of maximum pharmacophoric features and low toxicophoric and chemophoric features. Based on the comprehensive studies performed it is reasonable to state that the active compounds can be further developed as lead molecules against tuberculosis.| Properties | Compounds | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 4b | 4c | 4d | 4e | 4f | 4g | 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h | |
| a Computed using Screening Assistant (SA2) program. PDL (progressive drug like), PLL (progressive lead like).b Calculated using MOE (CCG) chemoinformatics suite.c PreADMET software.d LAZAR wherein Neg = negative and C = confidence value. | |||||||||||||||
| Lipinski rule of fivea | |||||||||||||||
| Molecular weight | 380.41 | 380.41 | 396.87 | 392.45 | 422.47 | 392.45 | 406.43 | 463.53 | 481.51 | 481.52 | 497.97 | 493.55 | 493.55 | 523.58 | 523.58 |
| HB acceptor | 4 | 4 | 4 | 5 | 6 | 5 | 6 | 5 | 5 | 5 | 5 | 6 | 6 | 7 | 7 |
| HB donor | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
5.442 | 5.442 | 5.881 | 5.247 | 5.238 | 5.284 | 4.996 | 4.911 | 5.064 | 5.062 | 5.501 | 4.904 | 4.867 | 4.858 | 4.858 |
|
|||||||||||||||
| Chemical properties | |||||||||||||||
| Weiner path b | 4228 | 4228 | 2152 | 2464 | 2912 | 2422 | 2664 | 3910 | 4284 | 4228 | 4228 | 4637 | 4693 | 5384 | 5332 |
| Ring counta | 4 | 4 | 4 | 4 | 4 | 4 | 5 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| PDL/PLLa | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
|
|||||||||||||||
| ADME properties | |||||||||||||||
| BBB (−3.0 to 1.2)c | 0.794 | 0.83 | 1.62 | 0.2 | 0.08 | 0.22 | 0.057 | 0.04 | 0.07 | 0.058 | 0.061 | 0.03 | 0.083 | 0.192 | 0.074 |
| Caco2 (nms) (<25, poor, >500, best)c | 31.67 | 30.74 | 24.03 | 37.38 | 41.81 | 21.94 | 24.79 | 33.27 | 31.81 | 31.33 | 24.31 | 33.43 | 36.71 | 39.38 | 37.02 |
| HIA (50–100%)c | 98.01 | 96.01 | 96.36 | 96.25 | 96.66 | 96.25 | 96.73 | 97.14 | 97.13 | 97.13 | 96.75 | 97.67 | 97.67 | 89.66 | 98.03 |
| Rotatable bonds (0–15)a | 3 | 3 | 3 | 4 | 5 | 4 | 3 | 4 | 4 | 4 | 4 | 5 | 5 | 6 | 6 |
| TPSA (7.0–200.0)b | 93.13 | 93.14 | 63.59 | 72.83 | 82.05 | 72.83 | 82.05 | 93.13 | 93.13 | 93.13 | 93.13 | 102.37 | 102.37 | 111.6 | 111.6 |
|
|||||||||||||||
| Toxicity propertiesd | |||||||||||||||
| DSSTox carcinogenic potency mutagenecity | Neg. (C: 0.129) | Neg. (C: 0.102) | Neg. (C: 0.115) | Neg. (C: 0.153) | Neg. (C: 0.153) | Neg. (C: 0.196 | Neg. (C: 0.129 | Neg.(C: 0.129 | Neg. (C: 0.169 | Neg. (C: 0.153) | Neg. (C: 0.129) | Neg. (C: 0.129) | Neg. (C: 0.129) | Neg. (C: 0.169) | Neg. (C: 0.169) |
| DSSTox carcinogenic potency mouse | Neg. (C: 0.080) | Neg. (C: 0.080) | Neg. (C: 0.011) | Neg. (C: 0.011) | Neg. (C: 0.011) | Neg. (C: 0.011) | Neg. (C: 0.085) | Neg. (C: 0.085) | Neg. (C: 0.085) | Neg. (C: 0.011) | Neg. (C: 0.085) | Neg. (C: 0.085) | Neg. (C: 0.085) | Neg. (C: 0.085) | Neg. (C: 0.085) |
QSAR modeling
We applied the QSAR approach to model the bioactivity of the chalcones in our quest to gather some clues regarding the molecular characteristics essential for biological activity. Initially, a simple 2D regression model was built using PLS approach for the series of chalcones using physico-chemical 2D structural descriptors implemented in MOE package26 which yielded a coefficient of determination 0.9449 and a root mean square error (RMSE) value of 1.70 (Fig. S1†). Next, we computed the 3D QM/QC based descriptors using PM3 and AM1 Hamiltonian for studying the electronic properties with an objective of mapping them with the variation in activity. The molecules were geometry optimized and subjected to energy minimization by employing the MMFF94x force field. The resultant 3D QSAR model yielded a R2 value of 0.87507, RMSE 2.72 and a cross validated R2(Q2) value of 0.687. The decrease in R squared value in the latter case can be attributed to the presence of few outliers such as compound 5b which showed better fit with 2D rather than 3D descriptors (Fig. S2†).For a comprehensive list of all the descriptors and their computed values, please refer to the ESI Table S1.† In the 3D model it was observed that the main properties correlating with the biological activity were the molecular orbital energies such as E(LUMO), E(HOMO). Frontier orbitals of valence electrons of compounds are known to play a major role in governing affinity towards biomolecules, specifically low values of LUMO indicate good electron acceptor tendency of the molecule and facilitate better interactions with the residue atoms in the active site of the receptor which eventually determine the biological activity. The molecules with lower E(LUMO) values were found to possess better biological activities (see entries 1, 9–11, Table S1†). All these molecules possess a halogen atom (X = F, Cl) on the benzene ring moiety. It is to be noted here that the docking studies also revealed the halogen substituted benzene ring to be involved in an arene cation type of interaction with the receptor residue atoms. The presence of a methoxy group in the benzene system on the other hand increased the E(LUMO) energies without affecting the E(HOMO) energies (entries 4–6) however when the piperidine group was present on the spiro skeleton, the methoxy group effect on the energies was not so prominent. It thus appears that the concurrent presence of halogen on the benzene ring moiety and piperidine on the spiro conjugated framework of the molecular system is essential for eliciting the bioactivity. A Genetic Programming based Symbolic Regression (GP-SR)27,28 modelling was performed to verify the correlation of electronic energy descriptors with bioactivity. The GP model produced a Correlation Coefficient (CC) value of 0.9757, 0.0684 (RMSE) and 1.7353 (MAPE). The GP expression obtained is shown in the eqn (1), parity plot of experimental versus GP predicted values is given in the ESI (Fig. S3†).
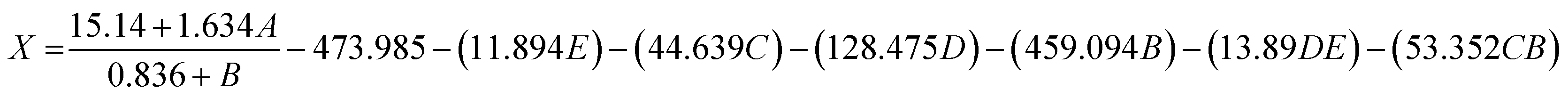 | (1) |
Thus the electronic energy descriptors based QSAR model developed can be employed in near future for virtual screening of not yet synthesized chalcone skeleton based molecules. To provide an in-depth analysis of molecular orbitals and derive more accurate energies, ab initio method was employed using DFT level of theory, B3LYP functional and 6-31G basis set. The HOMO LUMO energies in Gaussian 09 (ref. 29) were −6.92448 eV and −2.724675877 eV respectively. The iso density surface plots of HOMO and LUMO for the compound 5c possessing the best inhibitory activity are depicted in the Fig. 7. The HOMOs lie over the tricyclic spiro-chromone ring system while the LUMOs are located over the fluoro benzene ring moiety in the molecule.
 | ||
| Fig. 7 Iso density surface maps of frontier orbitals of molecule 5c with best inhibitory activity as determined in the in vitro Mtb assay. | ||
Conclusion
In summary, a series of spirochromone annulated chalcone conjugates were synthesized for the first time using an efficient synthetic procedure beginning from resorcinol. All these new compounds were characterized by 1H NMR, 13C NMR and mass spectral analysis. The in vitro antimycobacterial evaluation of all the synthesized compounds showed that five compounds possess moderate to good antimycobacterial activity. Noticeably, compound 5c is most potent compound in vitro with a MIC value of 3.13 μg mL−1, against MTB. Molecular docking studies were utilized to explore the putative targets among the known chalcone specific protein receptors. In theory the compounds showed greater affinity towards the mycobacterium phosphatase Ptp B enzyme, these studies will provide guidelines for future experimental efforts. Chemoinformatics analysis using a host of known and internally developed tools indicated that the compounds prioritized in this study possess good ADME profile, low toxicity and high pharmacophoric attributes essential for further development as anti-tubercular agents. Further, QSAR modeling established the correlation between frontier molecular orbital energies and biological activities of the compounds. The GP based expression can be used for screening new molecules for their therapeutic action.Experimental section
General methods
Solvents were purified and dried by standard procedures prior to use. 1H NMR and 13C NMR spectra were recorded on a Bruker AC-200 NMR spectrometer. Spectra were obtained in CDCl3. Monitoring of reactions was carried out using TLC plates Merck Silica gel 60 F254 and visualization with UV light (254 and 365 nm), I2 and anisaldehyde in ethanol as development reagents. HRMS (ESI) were recorded on a ORBITRAP mass analyser (Thermo Scientific, QExactive). Mass spectra were recorded at an ionization energy 70 eV on API Q Star Pulsar spectrometer using electrospray ionization. Ten fold serial dilutions of each test compound/drug were incorporated into Middlebrook 7H11 agar medium with OADC Growth Supplement. Inoculum of M. tuberculosis H37Rv were prepared from fresh Middlebrook 7H11 agar slants with OADC Growth Supplement adjusted to 1 mg mL−1 (wet weight) in Tween 80 (0.05%) saline diluted to 10−2 to give a concentration of approximately 107 cfu mL−1. A 5 μL amount of bacterial suspension was spotted into 7H11 agar tubes containing 10-fold serial dilutions of drugs per mL. The tubes were incubated at 37 °C, and final readings were recorded after 28 days. The minimum inhibitory concentration (MIC) is defined as the minimum concentration of compound required to give complete inhibition of bacterial growth.:5) to yield 3a (4.2 g, 62%) as a colourless solid. 1H NMR (200 MHz, CDCl3): δH = 1.44–1.75 (m, 8H), 1.94–2.03 (m, 2H), 2.63 (s, 3H), 2.70 (s, 2H), 6.48 (s, 1H), 8.36 (s, 1H), 12.82 (s, 1H); 13C NMR (50 MHz, CDCl3): δ 203.3 (CO), 190.5 (CO), 168.6 (C), 165.1 (C), 131.8 (CH), 115.0 (C), 113.9 (C), 105.1 (CH), 81.3 (C), 47.7 (CH2), 34.9 (CH2, 2 carbons), 26.4 (CH3), 24.9 (CH2), 21.3 (CH2, 2 carbons); MS: m/z 297 [M + Na]+.Synthesis of spirochromone annulated chalcone conjugates (4–5)
:25) to yield pure product 4–5. Spectroscopic data of compounds 4–5 are given below.
(E)-6-(3-(4-Fluorophenyl)acryloyl)-7-hydroxyspiro[chromane-2,1′-cyclohexan]-4-one (4a). Yellow solid; mp 156–157 °C, 1H NMR (200 MHz, CDCl3): δH = 1.46–1.77 (m, 8H), 1.97–2.05 (m, 2H), 2.73 (s, 2H), 6.50 (s, 1H), 6.95–7.19 (m, 2H), 7.51 (dd, J = 15.4, 9.3 Hz, 1H), 7.65–7.74 (m, 2H), 7.86 (dd, 15.4, 3.1 Hz, 1H), 13.5 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.3 (CO), 190.6 (CO), 169.9 (C), 165.2 (C), 161.8 (C), 144.4 (CH), 130.9 (CH, 2 carbons), 130.7 (CH), 130.6 (C), 119.2 (CH), 116.4 (CH, 2 carbons), 115.2 (C), 113.9 (C), 105.4 (CH), 81.4 (C), 47.8 (CH2), 35.0 (CH2, 2 carbons), 24.9 (CH2), 21.3 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C23H21FO4 [M + H]+ 381.1497, found 381.1495.
(E)-6-(3-(2-Fluorophenyl)acryloyl)-7-hydroxyspiro[chromane-2,1′-cyclohexan]-4-one (4b). Yellow solid; mp 188–189 °C; 1H NMR (200 MHz, CDCl3): δH = 1.46–1.77 (m, 8H), 1.97–2.03 (m, 2H), 2.73 (s, 2H), 6.50 (s, 1H), 6.95–7.24 (m, 2H), 7.38–7.49 (m, 1H), 7.69–7.82 (m, 2H), 8.03 (d, J = 15.4 Hz, 1H), 8.55 (s, 1H), 13.49 (s, 1H); 13C NMR (50 MHz, CDCl3): δC = 192.6 (CO), 190.6 (CO), 170.0 (C), 165.3 (C), 159.3 (C), 138.2 (CH), 132.4 (CH), 130.8 (CH), 129.5 (CH), 124.6 (CH), 122.0 (CH), 121.9 (C), 116.1 (CH), 115.3 (C), 114.0 (C), 105.4 (CH), 81.5 (C), 47.8 (CH2), 35.1 (CH2, 2 carbons), 25.0 (CH2), 21.4 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C23H21FO4 [M + H]+ 381.1497, found 381.1496.
(E)-6-(3-(2-Chlorophenyl)acryloyl)-7-hydroxyspiro[chromane-2,1′-cyclohexan]-4-one (4c). Yellow solid; mp 162–163 °C; 1H NMR (200 MHz, CDCl3): δH = 1.52–1.70 (m, 8H), 1.97–2.03 (m, 2H), 2.73 (s, 2H), 6.52 (s, 1H), 7.34–7.39 (m, 2H), 7.43–7.48 (m, 1H), 7.62 (d, J = 15.5 Hz, 1H), 7.82–7.86 (m, 1H), 8.30 (d, J = 15.5 Hz, 1H), 8.55 (s, 1H), 13.45 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.2 (CO), 190.6 (CO), 170.0 (C), 165.3 (C), 141.4 (CH), 135.8 (C), 132.7 (C), 131.6 (CH), 130.7 (CH), 130.3 (CH), 128.0 (CH), 127.1 (CH), 122.0 (CH), 115.2 (C), 113.9 (C), 105.4 (CH), 81.4 (C), 47.8 (CH2), 35.0 (CH2, 2 carbons), 24.9 (CH2), 21.3 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C23H21ClO4 [M + H]+ 397.1201, found 397.1203.
(E)-7-Hydroxy-6-(3-(4-methoxyphenyl)acryloyl)spiro[chromane-2,1′-cyclohexan]-4-one (4d). Yellow solid; mp 164–165 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47–1.67 (m, 8H), 1.96–2.03 (m, 2H), 2.72 (s, 2H), 3.87 (s, 3H), 6.50 (s, 1H), 6.94 (dd, J = 8.8, 2.8 Hz, 2H), 7.50 (d, J = 15.4 Hz, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.87 (dd, J = 15.2, 8.0 Hz, 1H), 8.55 (s, 1H), 13.73 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.5 (CO), 190.7 (CO), 170.0 (C), 165.0 (C), 162.1 (C), 145.7 (CH), 130.8 (CH, 2 carbons), 130.7 (CH), 127.1 (C), 116.8 (CH), 115.3 (C), 114.5 (CH, 2 carbons), 113.8 (C), 105.3 (CH), 81.3 (C), 55.4 (CH3), 47.8 (CH2), 35.0 (CH2, 2 carbons), 24.9 (CH2), 21.3 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C23H24O5 [M + H]+ 393.1697, found 393.1697.
(E)-6-(3-(2,4-Dimethoxyphenyl)acryloyl)-7-hydroxyspiro[chroma ne-2,1′-cyclohexan]-4-one (4e). Yellow solid; mp 215–216 °C; 1H NMR (200 MHz, CDCl3): δH = 1.51–1.77 (m, 8H), 1.97–2.03 (m, 2H), 2.71 (s, 2H), 3.88 (s, 3H), 3.93 (s, 3H), 6.48–6.49 (m, 2H), 6.54 (dd, J = 8.5, 2.4 Hz, 1H), 7.63 (d, J = 1.6 Hz, 1H), 7.67 (d, J = 8.6 Hz, 1H), 8.17 (d, J = 15.6 Hz, 1H), 8.56 (s, 1H), 13.88 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 193.1 (CO), 190.7 (CO), 170.1 (C), 164.9 (C), 163.6 (C), 160.7 (C), 141.4 (CH), 131.3 (CH), 130.5 (CH), 117.3 (CH), 116.7 (C), 115.6 (C), 113.7 (C), 105.6 (CH), 105.2 (CH), 98.4 (CH), 81.2 (C), 55.6 (CH3), 55.5 (CH3), 47.9 (CH2), 35.1 (CH2, 2 carbons), 25.0 (CH2), 21.4 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C25H26O6 [M + H]+ 423.1802, found 423.1801.
(E)-7-Hydroxy-6-(3-(3-methoxyphenyl)acryloyl)spiro[chromane-2,1′-cyclohexan]-4-one (4f). Yellow solid; mp 162–163 °C; 1H NMR (200 MHz, CDCl3): δH = 1.48–1.70 (m, 8H), 1.97–2.04 (m, 2H), 2.73 (s, 2H), 3.90 (s, 3H), 6.52 (s, 1H), 6.98–7.04 (m, 1H), 7.19–7.20 (m, 1H), 7.28–7.42 (m, 2H), 7.61 (d, J = 15.4 Hz, 1H), 7.87 (d, J = 15.4 Hz, 1H), 8.57 (s, 1H), 13.50 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.6 (CO), 190.6 (CO), 170.0 (C), 165.2 (C), 160.0 (C), 145.8 (CH), 130.7 (CH), 130.0 (CH), 121.5 (CH), 121.4 (C), 119.8 (CH), 117.1 (C), 117.0 (CH), 113.9 (C), 113.6 (CH), 105.4 (CH), 81.4 (C), 55.4 (CH3), 47.8 (CH2), 35.0 (CH2, 2 carbons), 25.0 (CH2), 21.3 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C25H24O5 [M + H]+ 393.1697, found 393.1703.
(E)-6-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-7-hydroxyspiro[chrom ane-2,1′-cyclohexan]-4-one (4g). Yellow solid; mp 162–163 °C; 1H NMR (200 MHz, CDCl3): δH = 1.52–1.77 (m, 8H), 1.97–2.03 (m, 2H), 2.73 (s, 2H), 6.06 (m, 2H), 6.51 (s, 1H), 6.85 (d, J = 7.9 Hz, 1H), 7.17 (dd, J = 8.2, 1.2 Hz, 1H), 7.23–7.24 (m, 1H), 7.46 (d, J = 15.5 Hz, 1H), 7.82 (d, J = 15.5 Hz, 1H), 8.54 (s, 1H), 13.66 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.4 (CO), 190.6 (CO), 170.0 (C), 165.1 (C), 150.4 (C), 148.5 (C), 145.6 (CH), 130.5 (CH), 128.8 (C), 125.8 (CH), 117.4 (CH), 115.3 (C), 113.8 (C), 108.7 (CH), 107.0 (CH), 105.3 (CH), 101.7 (CH2), 81.3 (C), 47.8 (CH2), 35.0 (CH2, 2 carbons), 24.9 (CH2), 21.3 (CH2, 2 carbons); HRMS (ESI): m/z calcd for C24H22O6 [M + H]+ 407.1489, found 407.1489.
tert-Butyl-6-cinnamoyl-7-hydroxy-4-oxospiro[chromane-2,4′-pip eridine]-1′-carboxylate (5a). Yellow solid; mp 165–166 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.61–1.69 (m, 2H), 1.99–2.04 (m, 2H), 2.75 (s, 2H), 3.22–3.24 (m, 2H), 3.87–3.96 (m, 2H), 6.54 (s, 1H), 7.46–7.47 (m, 3H), 7.66–7.72 (m, 3H), 7.94 (d, J = 15.5 Hz, 1H), 8.59 (s, 1H), 13.58 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.6 (CO), 189.6 (CO), 170.2 (C), 164.4 (C), 154.6 (CO), 146.1 (CH), 134.3 (C), 131.2 (CH), 130.8 (CH), 129.0 (CH, 2 carbons), 128.9 (CH, 2 carbons), 119.3 (CH), 115.7 (C), 113.7 (C), 105.5 (CH), 79.9 (C), 79.2 (C), 47.7 (CH2, 2 carbons), 47.6 (CH2), 34.2 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C27H29NO6 [M + Na]+ 486.1887, found 486.1897.
tert-Butyl-(E)-6-(3-(4-fluorophenyl)acryloyl)-7-hydroxy-4-oxospiro [chromane-2,4′-piperidine]-1′-carboxylate (5b). Yellow solid; mp 177–178 °C; 1H NMR (400 MHz, CDCl3): δH = 1.47 (s, 9H), 1.63–1.69 (m, 2H), 2.02 (bd, J = 13.7 Hz, 2H), 2.74 (s, 2H), 3.21–3.24 (m, 2H), 4.09–4.14 (m, 2H), 6.53 (s, 1H), 6.96 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 15.2 Hz, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.93 (d, J = 15.1 Hz, 1H), 8.57 (s, 1H), 13.77 (s, 1H); 13C NMR (100 MHz, CDCl3): δC 192.6 (CO), 189.7 (CO), 170.3 (C), 164.3 (C), 161.7 (C), 154.6 (CO), 146.2 (CH), 130.9 (CH, 2 carbons), 130.6 (C), 126.9 (CH), 116.6 (CH), 115.8 (C), 115.0 (C), 113.6 (CH, 2 carbons), 105.4 (CH), 79.9 (C), 79.1 (C), 47.7 (CH2, 3 carbons), 34.3 (CH2, 2 carbons), 28.4 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C27H28FNO6 [M + Na]+ 504.1793, found 504.1798.
tert-Butyl-(E)-6-(3-(2-fluorophenyl)acryloyl)-7-hydroxy-4-oxospiro[chromane-2,4′-piperidine]-1′-carboxylate (5c). Yellow solid; mp 174–175 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.63–1.69 (m, 2H), 2.01–2.05 (m, 2H), 2.75 (s, 2H), 3.21–3.24 (m, 2H), 3.89–3.90 (m, 2H), 6.54 (s, 1H), 7.14–7.25 (m, 2H), 7.42–7.47 (m, 1H), 7.71–7.78 (m, 2H), 8.06 (d, J = 15.4 Hz, 1H), 8.57 (s, 1H), 13.50 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.6 (CO), 189.5 (CO), 170.1 (C), 164.5 (C), 160.5 (C), 154.6 (CO), 138.5 (CH), 130.9 (C), 129.6 (CH), 124.6 (CH), 122.4 (C), 121.8 (CH), 116.4 (CH), 116.2 (CH) 115.6 (C), 113.7 (C), 105.5 (CH), 79.9 (C), 79.2 (C), 47.7 (CH2, 3 carbons), 34.3 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C27H28FNO6 [M + Na]+ 504.1793, found 504.1792.
tert-Butyl-(E)-6-(3-(2-chlorophenyl)acryloyl)-7-hydroxy-4-oxospiro [chromane-2,4′-piperidine]-1′-carboxylate (5d). Yellow solid; mp 183–184 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.58–1.73 (m, 2H), 2.00–2.05 (m, 2H), 2.75 (s, 2H), 3.17–3.29 (m, 2H), 3.88–3.93 (m, 2H), 6.55 (s, 1H), 7.36–7.50 (m, 3H), 7.61 (d, J = 15.5 Hz, 1H), 7.82–7.87 (m, 1H), 8.33 (d, J = 15.5 Hz, 1H), 8.57 (s, 1H), 13.49 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.3 (CO), 189.5 (CO), 170.1 (C), 164.5 (C), 159.3 (C), 154.6 (CO), 141.7 (CH), 135.8 (C), 132.6 (C), 131.8 (CH), 130.9 (CH), 130.4 (CH), 128.0 (CH), 127.2 (CH), 121.9 (CH), 113.7 (C), 105.5 (CH), 79.9 (C), 79.2 (C), 47.7 (CH2, 3 carbons), 34.3 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C27H28ClNO6 [M + Na]+ 520.1497, found 520.1500.
tert-Butyl-(E)-7-hydroxy-6-(3-(3-methoxyphenyl)acryloyl)-4-oxo spiro[chromane-2,4′-piperidine]-1′-carboxylate (5e). Yellow solid; mp 162–163 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.61–1.67 (m, 2H), 2.00–2.07 (m, 2H), 2.73 (s, 2H), 3.16–3.29 (m, 2H), 3.84–3.89 (m, 5H), 6.52 (s, 1H), 6.96–7.02 (m, 1H), 7.17–7.20 (m, 1H), 7.31–7.41 (m, 2H), 7.59 (d, J = 15.2 Hz, 1H), 7.87 (d, J = 15.2 Hz, 1H), 8.56 (s, 1H), 13.56 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.6 (CO), 189.5 (CO), 170.2 (C), 164.5 (C), 160.3 (C), 154.6 (CO), 146.2 (CH), 135.6 (CH), 130.8 (CH), 130.0 (CH), 121.5 (CH), 119.6 (C), 117.1 (CH), 115.7 (C), 113.7 (CH), 113.7 (C), 105.5 (CH), 79.9 (C), 79.2 (C), 55.4 (CH3), 47.7 (CH2, 3 carbons), 34.3 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C28H31NO7 [M + Na]+ 516.1993, found 516.2006.
tert-Butyl-(E)-7-hydroxy-6-(3-(4-methoxyphenyl)acryloyl)-4-oxo spiro[chromane-2,4′-piperidine]-1′-carboxylate (5f). Yellow solid; mp 176–177 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.60–1.73 (m, 2H), 2.00–2.06 (m, 2H), 2.74 (s, 2H), 3.18–3.29 (m, 2H), 3.89–3.90 (m, 5H), 6.53 (s, 1H), 6.95 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 15.7 Hz, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.89 (d, J = 15.7 Hz, 1H), 8.58 (s, 1H), 13.76 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 192.5 (CO), 189.7 (CO), 170.2 (C), 164.2 (C), 162.2 (C), 154.6 (CO), 146.0 (CH), 130.8 (CH, 2 carbons), 130.6 (CH), 127.0 (C), 116.7 (CH), 115.7 (C), 114.4 (CH, 2 carbons), 113.5 (C), 105.3 (CH), 79.9 (C), 79.0 (C), 55.4 (CH3), 47.6 (CH2, 3 carbons), 34.3 (CH2, 2 carbons), 28.4 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C28H31NO7 [M + H]+ 494.2173, found 494.2175.
tert-Butyl-(E)-6-(3-(2,4-dimethoxyphenyl)acryloyl)-7-hydroxy-4-oxospiro[chromane-2,4′-piperidine]-1′-carboxylate (5g). Yellow solid; mp 160–161 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.66–1.72 (m, 2H), 1.99–2.05 (m, 2H), 2.73 (s, 2H), 3.16–3.31 (m, 2H), 3.88–3.89 (m, 5H), 3.93 (s, 3H), 6.48–6.49 (m, 1H), 6.51 (s, 1H), 6.54–6.60 (m, 1H), 7.62–7.63 (m, 1H), 7.67 (d, J = 8.1 Hz, 1H), 8.18 (d, J = 15.4 Hz, 1H), 8.57 (s, 1H), 13.93 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 193.0 (CO), 189.7 (CO), 170.3 (C), 164.1 (C), 163.7 (C), 160.7 (C), 154.6 (CO), 141.7 (CH), 131.3 (CH), 130.6 (CH), 116.9 (CH), 116.5 (C), 115.9 (C), 113.5 (C), 105.5 (CH), 105.3 (CH), 98.3 (CH), 79.8 (C), 78.9 (C), 55.6 (CH3), 55.5 (CH3), 47.7 (CH2, 3 carbons), 34.2 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C29H33NO8 [M + Na]+ 546.2098, found 546.2126.
tert-Butyl-(E)-6-(3-(2,5-dimethoxyphenyl)acryloyl)-7-hydroxy-4-oxospiro[chromane-2,4′-piperidine]-1′-carboxylate (5h). Yellow solid; mp 150–151 °C; 1H NMR (200 MHz, CDCl3): δH = 1.47 (s, 9H), 1.70–1.72 (m, 2H), 2.00–2.07 (m, 2H), 2.74 (s, 2H), 3.16–3.29 (m, 2H), 3.84–3.86 (m, 5H), 3.91 (s, 3H), 6.53 (s, 1H), 6.88–7.03 (m, 2H), 7.19 (d, J = 3.0 Hz, 1H), 7.70 (d, J = 15.4 Hz, 1H), 8.21 (d, J = 15.4 Hz, 1H), 8.59 (s, 1H), 13.72 (s, 1H); 13C NMR (50 MHz, CDCl3): δC 193.0 (CO), 189.5 (CO), 170.1 (C), 164.3 (C), 154.5 (CO), 153.6 (C), 153.4 (C), 141.4 (CH), 130.8 (CH), 123.7 (C), 120.0 (CH), 118.2 (CH), 115.8 (C), 113.7 (CH), 113.6 (C), 112.4 (CH), 105.3 (CH), 79.8 (C), 79.0 (C), 56.0 (CH3), 55.9 (CH3), 47.7 (CH2), 47.6 (CH2, 2 carbons), 34.2 (CH2, 2 carbons), 28.3 (CH3, 3 carbons); HRMS (ESI): m/z calcd for C29H33NO8 [M + Na]+ 546.2098, found 546.2099.
Acknowledgements
Financial support from the CSIR Network projects (CSC0130, BSC0121 and CSC0108) is gratefully acknowledged. J. Robles acknowledges financial support from CONACYT (Research grant 168474). U. M. V. B. (378051/252139). E. D. C. acknowledges support from CONACYT graduate scholarship (368973/250842). L. C. B. acknowledges support from CONACYT graduate scholarship (327057/265238). RV thanks DST, New Delhi for the award of a women scientist fellowship (LS-201/2011). All the authors thank the anonymous referee for providing suggestions to improve the manuscript.Notes and references
- (a) Global Tuberculosis Report 2014, World Health Organization, Geneva, 2014 Search PubMed; (b) A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483 CrossRef CAS PubMed.
- (a) D. J. Newman and G. M. Cragg, in Natural Products in Medicinal Chemistry, ed. S. Hanessian, Wiley-VCH Verlag GmbH & Co. KgaA, 1st edn, 2014, part 1, pp. 1–41 Search PubMed; (b) S. Basu, B. Ellinger, S. Rizzo, C. Deraeve, M. Schurmann, H. Preut, H. D. Arndt and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6805 CrossRef CAS PubMed; (c) S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800 CrossRef CAS PubMed; (d) M. Manger, M. Scheck, H. Prinz, J. P. V. Kries, T. Langer, K. Saxena, H. Schwalbe, A. Furstner, J. Rademann and H. Waldmann, ChemBioChem, 2005, 6, 1749 CrossRef CAS PubMed.
- J. K. Lin and M. S. Weng, in The Science of Flavonoids, ed. E. Grotewold, Springer, New York, 2006, ch. 8, pp. 239–268 Search PubMed.
- D. N. Dhar, The Chemistry of Chalcones and Related Compounds, John Wiley & Sons, New York, 1981 Search PubMed.
- L. D. Chiaradia, A. Mascarello, M. Purificacao, J. Vernal, M. N. S. Cordeiro, M. E. Zenteno, A. Villarino, R. J. Nunes, R. A. Yunes and H. Terenzi, Bioorg. Med. Chem. Lett., 2008, 18, 6227 CrossRef CAS PubMed.
- (a) Y. M. Lin, PCT, WO 01/21164A2, 2001; (b) Y. M. Lin, Y. Zhou, M. T. Flavin, L. M. Zhou, W. Nie and F. C. Chen, Bioorg. Med. Chem., 2002, 10, 2795 CrossRef CAS PubMed; (c) Z. Nowakowska, Eur. J. Med. Chem., 2007, 42, 125 CrossRef CAS PubMed; (d) R. H. Hans, E. M. Guantai, C. Lategan, P. J. Smith, B. Wan, S. G. Franzblau, J. Gut, R. J. Rosenthal and K. Chibale, Bioorg. Med. Chem. Lett., 2010, 20, 942 CrossRef CAS PubMed.
- (a) L. D. Chiaradia, P. G. A. Martins, M. N. S. Cordeiro, R. V. C. Guido, G. Ecco, A. D. Andricopulo, R. A. Yunes, J. Vernal, R. J. Nunes and H. Terenzi, J. Med. Chem., 2012, 55, 390 CrossRef CAS PubMed; (b) A. Mascarello, M. Mori, L. D. Chiaradia-Delatorre, A. C. O. Menegatti, M. F. Delle, F. Ferrari, R. A. Yunes, R. J. Nunes, H. Terenzi, B. Botta and M. Botta, PLoS One, 2013, 8, e77081 CAS; (c) A. Mascarello, L. D. Chiaradia, J. Vernal, A. Villarino, R. V. C. Guido, P. Perizzolo, V. Poirier, D. Wong, P. G. A. Martins, R. J. Nunes, R. A. Yunes, A. D. Andricopulo, Y. Av-Gay and H. Terenzi, Bioorg. Med. Chem., 2010, 18, 3783 CrossRef CAS PubMed.
- B. W. Vanhoecke, F. Delporte, E. van Braeckel, A. Heyerick, H. T. Depypere, M. Nuytinck, D. de Keukeleire and M. E. Bracke, In Vivo, 2005, 19, 103 CAS.
- (a) M. Muthukrishnan, U. M. V. Basavanag and V. G. Puranik, Tetrahedron Lett., 2009, 50, 2643 CrossRef CAS; (b) M. Muthukrishnan and O. V. Singh, Synth. Commun., 2008, 38, 3875 CrossRef CAS; (c) M. Muthukrishnan, P. S. Patil, S. V. More and R. A. Joshi, Mendeleev Commun., 2005, 100 CrossRef CAS; (d) O. V. Singh, M. Muthukrishnan and R. Gopan, Synth. Commun., 2005, 35, 2723 CrossRef CAS; (e) O. V. Singh, M. Muthukrishnan and M. Sundaravadivelu, Indian J. Chem., 2005, 44B, 2575 CAS; (f) R. Vyas, M. Karthikeyan, G. Nainaru and M. Muthukrishnan, Comb. Chem. High Throughput Screening, 2015, 18, 624 CrossRef CAS.
- (a) M. Muthukrishnan, M. Mujahid, P. Yogeeswari and D. Sriram, Tetrahedron Lett., 2011, 52, 2387 CrossRef CAS; (b) M. Mujahid, R. G. Gonnade, P. Yogeeswari, D. Sriram and M. Muthukrishnan, Bioorg. Med. Chem. Lett., 2013, 23, 1416 CrossRef CAS PubMed.
- A. A. A. Emara and A. A. A. Abou-Hussen, Spectrochim. Acta, Part A, 2006, 64, 1010 CrossRef PubMed.
- ChemAxon Marvin Suite 15, accessed on 17th June 2015, https://www.chemaxon.com/download/marvin-suite/.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455 CAS.
- Chemical Computing Group Inc., Molecular Operating Environment Software, Montreal, 2010 Search PubMed.
- F. Macaev, V. Boldescu, S. Pogrebnoi and G. Duca, Med. Chem., 2014, 4, 487 CAS.
- C. Grundner, H. L. Ng and T. Alber, Structure, 2005, 13, 1625 CrossRef CAS PubMed.
- C. Grundner, D. Perrin, R. H. Van Huijsduijnen, D. Swinnen, J. Gonzalez, C. L. Gee and T. Alber, Structure, 2007, 15, 499 CrossRef CAS PubMed.
- K. A. Rawls, P. T. Lang, J. Takeuchi, S. Imamura, T. D. Baguley, C. Grundner, T. Alber and J. A. Ellman, Bioorg. Med. Chem. Lett., 2009, 19, 6851 CrossRef CAS PubMed.
- V. Le Guilloux, A. Arrault, L. Colliandre, S. Bourg, P. Vayer and L. Morin-Allory, J. Cheminf., 2012, 4, 20 CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337 CrossRef CAS.
- S. K. Lee, S. H. Park, I. H. Lee and K. T. No, PreAD-MET Ver. v2. 0, BMDRC, Seoul. Korea, 2007 Search PubMed.
- M. Tripathi, S. I. Khan, A. Thakur, P. Ponnan and D. S. Rawat, New J. Chem., 2015, 39, 3474 RSC.
- S. Ren and E. J. Lien, Caco-2 cell permeability vs. human gastro-intestinal absorption: QSPR analysis, in Progress in Drug Research, Birkhäuser, Basel, 2000, pp. 1–23 Search PubMed.
- A. Maunz, M. Gutlein, M. Rautenberg, D. Vorgrimmler, D. Gebele and C. Helma, Front. Pharmacol., 2013, 4, 38 Search PubMed.
- M. Karthikeyan, D. Pandit and R. Vyas, Comb. Chem. High Throughput Screening, 2015, 18, 544 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015.
- R. Vyas, P. Goel, M. Karthikeyan, S. S. Tambe and B. D. Kulkarni, Lett. Drug Des. Discovery, 2014, 11, 1112 CrossRef CAS.
- R. Vyas, P. Goel and S. Tambe, Applications of genetic programming in chemistry and chemical engineering, in Handbook of Genetic Programming applications, ed. A. H. Gandomi, A. H. Alavi and C. Ruan, Springer, 2015, ch. 5, in press, DOI:10.1007/978-3-319-20883-1-5.
- M. J. Frisch, et al., Gaussian 09, Revision A.01, Gaussian Inc Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1045946 (4f). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra21737g |
| This journal is © The Royal Society of Chemistry 2015 |