
Random site occupancy induced disordered Néel-type collinear spin alignment in heterovalent Zn2+–Ti4+ ion substituted CoFe2O4

Abstract
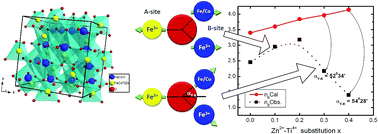
CoFe2O4, cobalt ferrite (CFO) nano-particles with composition CoZnxTixFe2−2xO4 (0 ≤ x ≤ 0.4) were synthesized by sol–gel autocombustion method. The effect of Zn2+–Ti4+ substitution on the structural, magnetic and frequency dependent permeability properties of the CFO nano-particles were investigated by X-ray diffraction, 57Fe Mössbauer spectroscopy, vibrating sample magnetometry, transmission electron microscopy and permeability analysis. The Rietveld refinement of XRD patterns confirm the single spinel phase and the crystallite size is found in the range of 22–32 nm. Cation distribution was estimated by refining the XRD pattern by Rietveld method, and shows Zn2+ ions at the tetrahedral A-sites, and Co2+ and Ti4+ ions at octahedral B-sites. The saturation magnetization (Ms) increased from 58 to 75 emu g−1 for up to x = 0.2 and then decreased, while the coercivity decreased continuously with Zn2+–Ti4+ substitution. Two distinct composition ranges with Zn2+–Ti4+ substitution are identified for which Ms variation with x is explained by the Néel and Yafet–Kittel models. The room temperature Mössbauer spectra are analyzed in detail for probing the magnetic properties of Fe based Zn2+–Ti4+ substituted CFO. The effect of Zn2+–Ti4+ substitution on various Mössbauer parameters, viz. hyperfine field distribution, isomer shift, quadrupole splitting, and line width, has also been studied. The variation of nuclear magnetic fields at the A and B sites is explained on the basis of A–B and B–B supertransferred hyperfine interactions. The CFO nanoparticle is considered to possess a fully inverse spinel structure with a Néel-type collinear spin alignment, whereas the Zn2+–Ti4+ substitution in CFO is found to be structurally and magnetically disordered due to the nearly random distribution of cations and the canted spin arrangement. This study also demonstrates that one can tailor the magnetic properties of CFO particles by optimizing the Zn2+–Ti4+ substitution. The increase in the permeability, saturation magnetization and lower loss factor makes the synthesized materials suitable for applications in microwave devices and deflection yokes.
 Please wait while we load your content...
Please wait while we load your content...

