DOI:
10.1039/C5RA21449A
(Paper)
RSC Adv., 2015,
5, 105643-105650
SnSb–ZnO composite materials as high performance anodes for lithium-ion batteries†
Received
15th October 2015
, Accepted 7th December 2015
First published on 8th December 2015
Abstract
SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anode materials were prepared by a chemical coprecipitation method. Their microstructures and morphologies were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM); the electrochemical performance was investigated by constant current charging–discharging, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The specific initial charge and discharge capacities of the synthesized SnSb–(ZnO)0.4 composite anode material were 801.6 mA h g−1 and 1064.6 mA h g−1, respectively, while the initial coulombic efficiency was 75.3%. The capacity remained at 751 mA h g−1 after 100 cycles and the capacity retention ratio was 92.5%, which demonstrated excellent electrochemical performance. The addition of ZnO significantly improved the electrochemical properties of the SnSb alloy, especially for the cycling stability and rate capability of the composite materials.
1 Introduction
Lithium ion batteries are used more and more widely since their successful development and have become power supplies for portable electronic products due to their advantages of high specific energy, wide application temperature range, low self-discharge rate and long cycling life. The anode material is one of the most important factors in improving energy density and cycling life of the battery and has attracted much attention of researchers worldwide. Compared with commercial carbon electrodes, alloy anode materials which have good conductivity, high theoretical capacity, and high rate capability are very promising alternatives for high energy lithium-ion batteries.
Alloys which were formed by lithium with many metals M (M = Mg, Ca, Al, Sn, Pb, As, Sb, Bi, Pt, Ag, Au, Zn, Cd, Hg) can be used as anode materials for lithium ion battery. For example, lithium alloying (or lithium insertion) materials such as Sn,1–8 SnO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9–11 and Sb12–16 have gained attention as potential candidates due to their high theoretical capacities and energy density. However, the large volume change of the anode materials during charge/discharge processes will easily cause cracking or pulverization, and consequently affect the cycling performance of electrode. An effective approach to solve this problem is to employ SnSb-based alloy as anode material,17–22 for example, SnSb–M (M = Cu, Ag, Ti, Co)23–28 alloy and SnSb/C29–34 composite electrodes in which all components will react with lithium but at different stages in the charge and discharge cycles. In such systems, when one component reacts with lithium, the other component can buffer the volume change of the reacting one, and therefore increase the mechanical stability of alloy electrodes. Although the initial irreversible capacity was improved to some extent, the cycling stability was still so poor that they are not available for commercialization.
9–11 and Sb12–16 have gained attention as potential candidates due to their high theoretical capacities and energy density. However, the large volume change of the anode materials during charge/discharge processes will easily cause cracking or pulverization, and consequently affect the cycling performance of electrode. An effective approach to solve this problem is to employ SnSb-based alloy as anode material,17–22 for example, SnSb–M (M = Cu, Ag, Ti, Co)23–28 alloy and SnSb/C29–34 composite electrodes in which all components will react with lithium but at different stages in the charge and discharge cycles. In such systems, when one component reacts with lithium, the other component can buffer the volume change of the reacting one, and therefore increase the mechanical stability of alloy electrodes. Although the initial irreversible capacity was improved to some extent, the cycling stability was still so poor that they are not available for commercialization.
Zinc oxide (ZnO) was one of the most promising candidates for anode materials due to its high theoretical capacity, low cost, nature abundance, and no toxicity.35 However, its poor cyclability caused by volume expansion during the charge/discharge affects the practical application. In this work, for the first time, ZnO was introduced to the binary SnSb materials by chemical reduction. ZnO with zero dimensional spherical structure was able to effectively relieve the agglomeration of the SnSb alloy particle. And its intercalation potential was relatively low at about 0.2 V, which could play an important role in supporting lithium insertion/extraction, preventing the loss of electric contact between the current collector and electrode materials induced by the volume expansion. In addition, ZnO has a high theoretical capacity of 978 mA h g−1, which could improve the specific capacity of the composite materials. The electrochemical performance of SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anodes was studied and the dependence of the cyclic behavior of the electrodes and their microstructures was discussed. Especially, the specific initial charge and discharge capacities of the synthesized SnSb–(ZnO)0.4 composite anode material were 801.6 mA h g−1 and 1064.6 mA h g−1, respectively, while the initial coulombic efficiency was 75.3%. The capacity remained at 751 mA h g−1 after 100 cycles and the capacity retention ratio was 92.5%, which demonstrated excellent electrochemical performance.
2 Experimental
2.1 Synthesis of materials
All reagents used were of analytical quality. Nano-sized SnSb–(ZnO)x (x = 0,0.1, 0.2, 0.4, 0.6, 0.8) composite materials were synthesized by chemical reduction. 0.1 M of SbCl3, SnCl2, Zn(NO3)2 and C6H5Na3O7 with the molar ratio of 1:1:x:3.5 (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) as well as a 0.2 M aqueous solution of NaBH4 (pH > 12) were prepared, respectively. Excess amount of NaBH4 was added dropwise to the SbCl3/SnCl2/Zn(NO3)2 solution with strong magnetic stirring at room temperature. To obtain a better microstructure, all reduced powders were aged in an water bath at a constant temperature of 80 °C for 5 h. The precipitates were collected by centrifugation, washed with distilled water and ethanol, and dryed. Finally, the products were passed through a 200 mesh sieve and stored in a glove box under Ar.
2.2 Physical characterisations
The phases of the composite materials were analyzed using a D8 Advance X-ray diffraction (XRD) instrument. The microstructure and morphology of the particles were observed using field-emission scanning electron microscopy (FE-SEM) by a Hitachi Limited S-3400N instrument. Surface characterization for chemically reacted and electrochemically cycled electrodes was carried out with ex situ FTIR spectroscopy, using IR spectrometer (Spectrum One Version B).
2.3 Electrochemical characterizations
To evaluate their electrochemical properties, electrodes were prepared by 70 wt% active materials, 10 wt% acetylene black and 20 wt% carboxyl methyl cellulose (CMC) and styrene butadiene rubber (SBR) to form a slurry, which was then pressed onto a copper grid and dried at 120 °C for 10 h under vacuum. The loading of active materials was about 1.2 mg cm−2 on the electrodes.
The electrochemical performances was measured using coin cells assembled in an argon-filled glove box; these cells contained metallic lithium foil as a counter electrode, 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC) (1:1 v/v) as the electrolyte, Celgard 2400 membrane as a separator and the synthesized composite alloy as a working electrode. The galvanostatic charge–discharge performance was assessed using a CT2001A battery tester from 0.1 to 2.0 V (vs. Li/Li+) at a constant current density of 0.1 C at room temperature unless noted. Cyclic voltammetry (CV) from 0 to 2.0 V (vs. Li/Li+) at 0.02 mV s−1 and electrochemical impedance spectroscopy (EIS) in the frequency range 100 MHz to 0.01 Hz and with an amplitude of 5 mV were performed using a Solartron 1470e electrochemical testing system.
3 Result and discussion
3.1 Structures and morphologies analysis
Fig. 1a–g show the SEM images of SnSb, ZnO and SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite materials. As shown in Fig. 1f and g, the size of SnSb alloy particles is relatively small with a uniform distribution, while the obtained ZnO shows a spherical porous morphology, which could play a role in preventing the reunion and reducing the mechanical stress between the particles. The SnSb–(ZnO)x samples were formed by tiny irregular particles and aggregated into homogenous secondary particles; the particle size of the nano-composite materials was about 100 nm. In addition, with the increase of ZnO content, the particle size increased. Fig. 1h shows the X-ray diffraction (XRD) patterns of SnSb, ZnO and SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite materials obtained by chemical reduction method. The diffraction patterns of the composites contain sharp peaks for ZnO, SnSb and Sn and weak peaks for SnOx and SbOx. SnSb alloy mainly consists of a rhombohedral structure of the β-SnSb phase, in which there is a non-stoichiometric compound in the β-SnSb phase.27 Sn will be in excess, resulting in forming pure Sn crystals. As the amount of ZnO increased, the peaks for SnSb and Sn gradually weakened, while the peaks for ZnO were increased.
 |
| | Fig. 1 SEM images of (a) SnSb–(ZnO)0.1; (b) SnSb–(ZnO)0.2; (c) SnSb–(ZnO)0.4; (d) SnSb–(ZnO)0.6; (e) SnSb–(ZnO)0.8; (f) SnSb; (g) ZnO; and (h) XRD patterns of the materials. | |
3.2 Electrochemical performance of SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anode materials
Cycling performances of SnSb, ZnO and SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anode materials were investigated by galvanostatic charge/discharge between 0–2.0 V at a constant current density of 0.1 mA cm−2; the results are shown in Fig. 2a. They indicate that the discharge capacity of SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composites for the first cycle are 1260.4, 1489.4, 1729.5, 1064.6, 937 and 905 mA h g−1, respectively, which far exceed the theoretical capacity of graphite. During the synthesis procedure, Sn and Sb oxides were inevitably generated, which would react with lithium, therefore improving the initial capacities. The reactions of metal oxide compounds with lithium are irreversible, so the irreversible capacity for the first cycle is also increased. The discharge capacities for the second cycle were 981.5, 1151.8, 1257.9, 883.3, 801.2 and 688.6 mA h g−1, and the capacity stayed at 405.3, 551, 453, 739, 680 and 597 mA h g−1 after 100 cycles. The capacities for the SnSb and ZnO after 100 cycles were 401.4 and 193.6 mA h g−1, respectively. Compared to the former, the SnSb–ZnO0.4 composite materials exhibited greatly improved cycling stability with a discharge capacity of 739 mA h g−1 after 100 cycles and capacity retention of 92% compared to the 2nd cycle (a discharge capacity of 883.3 mA h g−1). The average capacity attenuation rate was 0.08% from the 2nd to the 100th cycle. Therefore, SnSb alloy played an important role in the electrochemical performance. Both Sn (Li4.4Sn 990 mA h g−1) and Sb (Li3Sb 660 mA h g−1) improved the overall specific capacity, and due to the difference in electrode potential, the lithium insertion/extraction reactions will proceed in a stepwise manner. Moreover, it was demonstrated that with the addition of ZnO, the electrochemical performance of SnSb–(ZnO)x was significantly improved. The reason is that ZnO has a high theoretical capacity of 978 mA h g−1, which could improve the specific capacity of the composite materials. In addition, ZnO with zero dimensional spherical morphology was able to effectively relieve the agglomeration of the SnSb alloy particles. For example, the porosisy analysis results for SnSb and SnSb–(ZnO)0.4 are shown Fig. S1 (ESI†), the distribution of pores is more uniform for the composite material and the pore ratio for SnSb and SnSb–(ZnO)0.4 are 11.1% and 20.8%, respectively. The intercalation potential for ZnO was relatively low at about 0.2 V, which played a role in supporting the process of lithium insertion/extraction, preventing the lost of electric contact between the current collector and electrode materials induced by volume expansion. Fig. 2b shows the curves of initial discharge and charge profiles of SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anodes at 0.01 mA cm−2 over an operating voltage of 0–2.0 V. The trends of the curves are similar, indicating that ZnO contents hardly affect the mechanism of lithium extraction and insertion for composites. The initial discharge curves are divided into three parts with two main discharge plateaus. The first part shows a rapidly decrease over 0.8 V, corresponding to irreversible reaction such as the reduction of oxides, the electrolyte decomposition and the formation of solid electrolyte interface (SEI) film. The SEI film on the active particles prevented them from reacting with the electrolyte subsequent, which reduced the irreversible capacity in subsequent reactions. A plateau appears from 0.8 to 0.65 V, which is likely due to the reduction of tin oxides and the reaction of SnSb with lithium. The content of the SnSb is reduced as the ZnO amount increases in the electrodes, and this resulted in a narrower plateau at 0.8 V. The third part of the curve is from 0.5–0 V, which correspond to both the initial and the extruded Sn reacting with Li to form various LixSn compounds. However, with the addition of ZnO, the voltage curves gradually became smooth and continuous. The plateau at approximately 0.2 V corresponds to the lithium intercalation process into ZnO.
 |
| | Fig. 2 (a) Cycling performances, (b) voltage profiles (c) rate capabilities of SnSb, ZnO and SnSb–(ZnO)x composite materials, and (d) electrochemical performance of SnSb–ZnO0.4 at 0.1 mA cm−2. | |
Fig. 2c shows the specific capacities of the ZnO, SnSb and SnSb–(ZnO)0.4 electrodes at different charge/discharge rates. The SnSb–(ZnO)0.4 composite material exhibited excellent cycling stability at different charge/discharge rates. During the preceding 60 cycles, the anodes discharged at increasing current densities. The average specific capacities decreased as the current density increased, and the initial discharge capacities of ZnO, SnSb and SnSb–(ZnO)0.4 are 1365.2, 1330.2 and 1187.3 mA h g−1, with the coulombic efficiency are 60.8%, 77.9% and 83.3%, respectively. The anode materials became fully lithiated with a low current density, and the reactions were nearly balanced, therefore exhibiting high capacities. At high current densities, the ions in the liquid phase diffused quickly and most of the Li+ rapidly arrived at the surface of the electrodes, resulting in different equilibrium rates in the reactions, while the capacities decreased. However, even at 2.5 mA cm−2, the discharge capacity of SnSb–(ZnO)0.4 composite anode material still reached 552 mA h g−1, which is far higher than the theoretical capacity of graphite. The addition of ZnO not only increased the discharge capacity, but also prevented the agglomeration of alloy nanoparticles. When the current density varied from 2.5 to 0.05 mA cm−2, the discharge specific capacities increased gradually; the discharge capacities of SnSb–(ZnO)0.4 recovered to 854.8 mA h g−1, with a capacity retention rate of 92.5%, showing excellence rate capability.
To confirm the cycling stability of the SnSb–ZnO0.4 composite material at high current density, the electrochemical performance was evaluated at 0.1 mA cm−2 in the voltage range of 0–2.0 V. As shown in Fig. 2d, the initial discharge capacity of SnSb–ZnO0.4 composite material was 1064.6 mA h g−1 and the coulombic efficiency of 75.3%. Nevertheless, the coulombic efficiency of the 3rd and later cycles all exceeded 94%, reaching 98% after 10th cycle. The capacity remained at 751 mA h g−1 after 100 cycles and the capacity retention ratio was 92.5%, showing excellence cycle stability.
Fig. 3 shows the cycling performance of SnSb–ZnO0.4 over different operating voltage ranges. When the cutoff voltages were 1.5 and 2.0 V, the initial discharge capacities were 1001.5 and 1064.6 mA h g−1, the second discharge capacities were 801.2 and 883.3 mA h g−1, and the capacity remained at 542.7 and 739 mA h g−1 after 100 cycles, while the capacity retention ratio was 67.7% and 83.7%, respectively. The results indicated that the initial discharge capacity increased as the cut off voltage increased, meanwhile the cycling stability also improved. When the cutoff was 1.5 V, the composite material was not completely reacting with lithium, resulting in lower utilization of materials and lower initial discharge capacity. While the cutoff voltage was 2 V, the anodes exhibited a higher capacity for deep lithiation in the first cycle. As the cutoff voltage for charge increased, particles can reacted completely with lithium, which increased the amount of active particles within subsequent cycles, and therefore improved the electrochemical properties. The relationship between the lithiation depth and the operating voltage greatly affected the charge/discharge capacities. It was reported that for SnSb alloy, the reaction between Li3Sb and SnSb is prohibited and the Li3Sb can be acted as an inert phase in whole reaction process in lower cut-off potential range (0.02–0.9 V), while the volume and structure changes were alleviated effectively and the excellent cycling performance with the reversible capacities was obtained.16 However, the electrochemical properties can be improved by increasing the upper cutoff voltage after the introduction of ZnO in our study, which not only increase the depth of lithium insertion/extraction, but also relieve the volume expansion and restrain the destruction of the inner stress, boosting the lithiation efficiency.
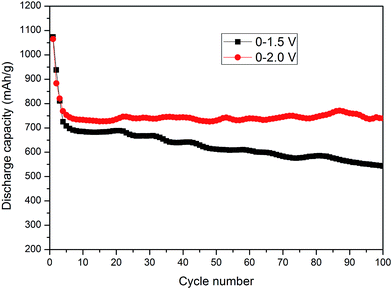 |
| | Fig. 3 Cycling performance of SnSb–(ZnO)0.4 at different operating voltage ranges at 0.01 mA cm−2. | |
Cyclic voltammograms of SnSb and SnSb–(ZnO)0.4 composite material were tested in the voltage range of 0–2.0 V (vs. Li/Li+) at a scanning rate of 0.02 mV s−1. As shown in Fig. 4, there is almost no change in peak shape for the 2nd and 3rd cycles, exhibiting a high reversibility. And the relatively large difference between the SnSb and SnSb–(ZnO)0.4 is due to the participation of ZnO with the lithium intercalation reactions. For the cathodic scan, a low reduction peak is observed in the first cycle at 1.5 to 0.8 V, but this peak disappears in subsequent cycles, corresponding to the reduction of the oxides, the decomposition of the electrolyte and the formation of SEI film. A strong reduction peak appeared at approximately 0.8 V and persisted through subsequent cycles, which corresponded to Sb in the SnSb alloy reacting with Li to form Li3Sb, while the Sn was released in its elemental form. Afterwards, a series of peaks appeared after 0.66 V, corresponding to the lithiation of Sn to form a series of LixSn alloys, the reaction of ZnO with Li to form LiZn, etc. In the anodic scan, a series of weak peaks appeared at 0.4 V, corresponding to the delithiation of LixSn and the strong peaks at 0.8 and 1.1 V were attributed to the delithiation of LixSb and the formation of SnSb alloys, respectively.
 |
| | Fig. 4 CV curves of (a) SnSb and (b) SnSb–(ZnO)0.4 composite materials at a scanning rate of 0.02 mV s−1. | |
Overall, the lithiation mechanisms for SnSb alloys are:
| | |
3Li + SnSb → Li3Sb + Sn
| (1) |
| | |
xLi + Sn → LixSn (x ≤ 4.4)
| (2) |
and the lithiation mechanisms for SnSb–(ZnO)
x composites are:
| | |
3Li + SnSb → Li3Sb + Sn
| (3) |
| | |
xLi + Sn → LixSn (x ≤ 4.4)
| (4) |
| | |
2Li + ZnO → Zn + Li2O
| (5) |
The solid electrolyte interface (SEI) formation may be a significant factor that influences the cyclability of the battery. Fig. 5 shows the FTIR spectra of the surface layer of the SnSb and SnSb–(ZnO)0.4 electrodes after discharge for 100 cycles. The peaks at ∼855 cm−1 are attributed to stretching modes of P–F.36 A small peak at 1649 cm−1 is a characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl group of R–CO2−Mn+ (R = alkyl group) carboxylate metal salt.37 The peaks at 1071 and 1219 cm−1 attributed to C–O, C–C–O and C–O–O group confirm this assignment. Two peaks in the region of 2950–2840 cm−1 are attributed to stretching modes of CH3– and –CH2– indicating the presence of CH3CH2-alkyl group.38 The peak at 1509 cm−1 is assigned to CO stretching modes of Li2CO3, which is a common component of the SEI layer.39 The peak intensity of Li2CO3 for SnSb is stronger than that for SnSb–(ZnO)0.4, indicating more accumulated SEI layer was produced. Moreover, the relative weak peaks for CH3CH2-alkyl group and CO carbonyl group (R–CO2−Mn+) suggest the conversion of the species to Li2CO3.40 This may be one reason that SnSb showed lower cycling performance because of low conductivity of Li2CO3 and the detailed mechanism is under study.
O carbonyl group of R–CO2−Mn+ (R = alkyl group) carboxylate metal salt.37 The peaks at 1071 and 1219 cm−1 attributed to C–O, C–C–O and C–O–O group confirm this assignment. Two peaks in the region of 2950–2840 cm−1 are attributed to stretching modes of CH3– and –CH2– indicating the presence of CH3CH2-alkyl group.38 The peak at 1509 cm−1 is assigned to CO stretching modes of Li2CO3, which is a common component of the SEI layer.39 The peak intensity of Li2CO3 for SnSb is stronger than that for SnSb–(ZnO)0.4, indicating more accumulated SEI layer was produced. Moreover, the relative weak peaks for CH3CH2-alkyl group and CO carbonyl group (R–CO2−Mn+) suggest the conversion of the species to Li2CO3.40 This may be one reason that SnSb showed lower cycling performance because of low conductivity of Li2CO3 and the detailed mechanism is under study.
 |
| | Fig. 5 FTIR spectra for SnSb and SnSb–(ZnO)0.4 electrodes after discharge for 100 cycles. | |
3.3 The relationship between structure and electrochemical performances
The microstructures of SnSb–(ZnO)0.4 before and after cycling are shown in Fig. 6. As shown in Fig. 6a, the morphologies and microstructures of anodes show a porous feature, and the active materials were separately dispersed. After 10 cycles, the particles could not be observed because SEI films formed constantly during cycling and the particles size continued to decrease. Meanwhile, a significant volume expansion occurred while lithiating the alloy particles, the aggregate expanded around the area when physical space was unavailable, as shown in Fig. 6b. Details regarding the microstructure of SnSb–(ZnO)0.4 during cycling are shown in Fig. 6c and d. The microstructures of the electrodes changed dramatically after 70 and 100 cycles. There were obvious cracks on the surface of electrodes and the active material broke off from the copper foil.
 |
| | Fig. 6 SEM images of the SnSb–(ZnO)0.4 composite anode materials: after (a) 0, (b) 10, (c) 70 and (d) 100 cycles. | |
Fig. 7a compares the electrochemical impedance diagrams of SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anode materials and the fitting curves before cycling at the open circuit voltage; they are composed of an arc with a large radius and a sloping line. The radius increases as the amount of ZnO increases, and the high-frequency intercept at the Z-axis and the slope of the linear component at low-frequency show little change. The semicircular arc of the high-frequency region at this time represented the contact resistance of electrode materials. The inset of Fig. 7b shows the impedance responses, which were then analyzed using an equivalent circuit that takes into account all possible contributions to the impedance of the test cell, using resistance Rs as the electrolyte impedance, Qg as the diffusive resistance and Qcf and Rcf as the contact resistances. All of the equivalent circuits were fit very well with the experimental data, and the representative fitting results are shown in Table 1. With the addition of ZnO which has zero dimensional spherical structure, the size of micropore increases and the electrolyte can fully penetrate into the alloy particles to form a SEI film which is conductive for lithium-ions.
 |
| | Fig. 7 EIS of (a) SnSb–(ZnO)x electrodes at the open circuit voltage, (b) SnSb and SnSb–(ZnO)0.4 electrodes after different cycles. | |
Table 1 Impedance parameters of the SnSb–(ZnO)x composite materials before cycling at the open circuit voltage
| SnSb–(ZnO)x |
0 |
0.1 |
0.2 |
0.4 |
0.6 |
0.8 |
| Rs (Ω) |
2.75 |
3.75 |
3.79 |
4.12 |
6.25 |
7.77 |
| Rct (Ω) |
88.07 |
182.76 |
242.09 |
477.31 |
586.49 |
713.06 |
Fig. 7b shows a comparison of the electrochemical impedance response and the fitting curves of SnSb and SnSb–(ZnO)0.4 composite anode materials after different cycles. And the representative fitting results are shown in Table S1.† As can be seen, the internal resistance was increased with the cycling. These results were mainly attributed to the collapse of the structure and the blocked transmission channels after continuous powdering and shredding of the active materials. During the following cycles, the resistance of the SnSb–(ZnO)0.4 composite anode material is much lower than that of the SnSb alloy, which exhibited a more stable structure. The shredded powders covered on the surface, prevented the Li+ from being trapped into the active materials and then produced an increased interfacial resistance. Ultimately, the total impedance increased after the cycles.
4 Conclusions
SnSb–(ZnO)x (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8) composite anode materials were prepared by a chemical coprecipitation method. The materials exhibited superior capacity and good cycling stability. ZnO with zero-dimensional spherical structure was able to effectively relieve the agglomeration of the SnSb alloy particles. This approach greatly shortened the lithium ion diffusion distance, reduced the trapping of lithium in the active materials, and increased the space available for volume expansions, resulting in greatly improved cycling stability for the SnSb–(ZnO)x composite anode materials. When the molar ratio of ZnO was 0.4 in the voltage ranging from 0 to 2.0 V, the initial specific charge capacity and discharge capacity of the synthesized SnSb–(ZnO)0.4 composite anode material were 801.6 mA h g−1 and 1064.6 mA h g−1, respectively, with an initial coulombic efficiency of 75.3%. Meanwhile, adjusting the voltage range by controlling the lithiation of ZnO and preventing the loss of contact between the particles and the copper foil resulted in excellent cycling performance for SnSb–(ZnO)x composite anode materials. When the cutoff voltage was 0–2.0 V, the discharge capacity remained at 751 mA h g−1 after 100 cycles and the capacity retention ratio was 92.5%, which is excellent electrochemical performance. This study gives a rational direction for designing the components and constructing the structures of composite anode materials to improve the performance of lithium ion batteries.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51374146), the Natural Science Foundation of Guangdong (2015A030313542), the Shenzhen Dedicated Funding of Strategic Emerging Industry Development Funds (JCYJ20130329113849606, JCYJ20140418182819155) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars.
References
- Z. Shen, Y. Hu, Y. L. Chen, X. W. Zhang, K. H. Wang and R. Z. Chen, J. Power Sources, 2015, 278, 660–667 CrossRef CAS.
- P. Nithyadharseni, M. V. Reddy, B. Nalini, M. Kalpana and B. V. R. Chowdari, Electrochim. Acta, 2015, 161, 261–268 CrossRef CAS.
- Y. Zhong, X. Li, Y. Zhang, R. Li, M. Cai and X. Sun, Appl. Surf. Sci., 2015, 332, 192–197 CrossRef CAS.
- Z. Yang, A. A. Gewirth and L. Trahey, ACS Appl. Mater. Interfaces, 2015, 7, 6557–6566 CAS.
- J. Zhu, L. Chen, Z. Xu and Bi. Lu, Nano Energy, 2015, 12, 339–346 CrossRef CAS.
- Y. Zhao, X. Li, B. Yan, D. Li, S. Lawes and X. Sun, J. Power Sources, 2015, 274, 869–884 CrossRef CAS.
- X. Li, A. Dhanabalan, L. Gu and C. Wang, Adv. Energy Mater., 2012, 2, 238–244 CrossRef CAS.
- X. Li, Y. Zhong, M. Cai, M. Balogh, D. Wang, Y. Zhang, R. Li and X. Sun, Electrochim. Acta, 2013, 89, 387–393 CrossRef CAS.
- T. Yang and B. Lu, Phys. Chem. Chem. Phys., 2014, 16, 4115–4121 RSC.
- W. Shi and B. Lu, Electrochim. Acta, 2014, 133, 247–253 CrossRef CAS.
- J. Zhu, G. Zhang, X. Yu, Q. Li, B. Lu and Z. Xu, Nano Energy, 2014, 3, 80–87 CrossRef CAS.
- J. H. Sung and C.-M. Park, J. Electroanal. Chem., 2013, 700, 12–16 CrossRef CAS.
- M. Saubanere, M. Ben Yahia, F. Lemoigno and M.-L. Doublet, J. Power Sources, 2015, 280, 695–702 CrossRef CAS.
- N. Li, S. Liao, Y. Sun, H. W. Song and C. X. Wang, J. Mater. Chem. A, 2015, 3, 5820–5828 CAS.
- L. Fan, J. Zhang, J. Cui, Y. Zhu, J. Liang, L. Wang and Y. Qian, J. Mater. Chem. A, 2015, 3, 3276–3280 CAS.
- F. Wang, M. Zhao and X. Song, J. Alloys Compd., 2009, 472, 55–58 CrossRef CAS.
- P. Nithyadharseni, M. Reddy, B. Nalini and B. Chowdari, Mater. Lett., 2015, 150, 24–27 CrossRef CAS.
- S.-C. Chao, Y.-F. Song, C.-C. Wang, H.-S. Sheu, H.-C. Wu and N.-L. Wu, J. Phys. Chem. C, 2011, 115, 22040–22047 CAS.
- L. Xue, X. Xia, T. Tucker, K. Fu, S. Zhang, S. Li and X. Zhang, J. Mater. Chem. A, 2013, 1, 13807–13813 CAS.
- F.-S. Ke, L. Huang, L. Jamison, L.-J. Xue, G.-Z. Wei, J.-T. Li, X.-D. Zhou and S.-G. Sun, Nano Energy, 2013, 2, 595–603 CrossRef CAS.
- L. Baggetto, H.-Y. Hah, J.-C. Jumas, C. E. Johnson, J. A. Johnson, J. K. Keum, C. A. Bridges and G.
M. Veith, J. Power Sources, 2014, 267, 329–336 CrossRef CAS.
- Q. Jiang, M. Jia, R. Xue and X. Shi, J. Power Sources, 2013, 37, 200–203 CAS.
- M. Zhao, Q. Zheng, F. Wang, Y. Qin and X. Song, J. Nanosci. Nanotechnol., 2010, 10, 7025–7030 CrossRef CAS PubMed.
- Q. Jiang, R. Xue and M. Jia, Appl. Surf. Sci., 2012, 258, 3854–3858 CrossRef CAS.
- X.-J. Pang, C.-H. Tan, X.-H. Dai, X. Wang, G.-W. Qi and S.-Y. Zhang, J. Appl. Electrochem., 2015, 45, 115–122 CrossRef CAS.
- C. Marino, M. T. Sougrati, A. Darwiche, J. Fullenwarth, B. Fraisse, J. C. Jumas and L. Monconduit, J. Power Sources, 2013, 244, 736–741 CrossRef CAS.
- Y. Wang, P. Zhang, X. Ren and G. Yi, J. Electrochem. Soc., 2011, 158, A1404–A1410 CrossRef CAS.
- C. Nithya, T. Sowmiya, K. Vijaya Baskar, N. Selvaganeshan, T. Kalaiyarasi and S. Gopukumar, Solid State Sci., 2013, 19, 144–149 CrossRef CAS.
- L. Fan, J. Zhang, Y. Zhu, X. Zhu, J. Liang, L. Wang and Y. Qian, RSC Adv., 2014, 4, 62301–62307 RSC.
- S. Fan, T. Sun, X. Rui, Q. Yan and H. H. Hng, J. Power Sources, 2012, 201, 288–293 CrossRef CAS.
- Z. Peixin, W. Yanyi, R. Xiangzhong, L. Kun and C. Shiguo, J. Power Sources, 2013, 233, 166–173 CrossRef.
- J. Li, Q. Ru, S. Hu, D. Sun, B. Zhang and X. Hou, Electrochim. Acta, 2013, 113, 505–513 CrossRef CAS.
- K. H. Seng, Z. P. Guo, Z. X. Chen and H. K. Liu, Adv. Sci. Lett., 2011, 4, 18–23 CrossRef CAS.
- J. Li, Q. Ru, S.-J. Hu and L.-Y. Guo, Acta Phys. Sin., 2014, 63, 168201 Search PubMed.
- L. Fan, J. Zhang, Y. Zhu and Y. Qian, Mater. Chem., 2014, 59, 2006–2011 CAS.
- S. Song, R. Reade, E. Cairns, J. Vaughey, M. Thackeray and K. Striebel, J. Electrochem. Soc., 2004, 151, A1012–A1019 CrossRef CAS.
- G. Zhuang, H. Yang, P. Ross Jr, K. Xu and T. Jow, Electrochem. Solid-State Lett., 2005, 9, A64–A68 CrossRef.
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electrochem. Soc., 1987, 134, 1611–1620 CrossRef CAS.
- K. Kanamura, H. Tamura and Z. Takehara, J. Electroanal. Chem., 1995, 394, 49–62 CrossRef.
- S. Song and S. Baek, Electrochim. Acta, 2009, 54, 1312–1318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21449a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






