
Direct, rapid, solvent-free conversion of unactivated esters to amides using lithium hydroxide as a catalyst†
Abstract
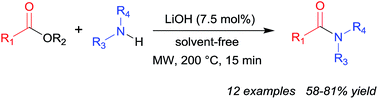
A simple, solvent-free methodology is reported for the direct conversion of esters to amides using lithium hydroxide as a catalyst. The approach allows for the preparation of a range of amide products as well as being applicable to the ring-opening of a representative lactone.
 Please wait while we load your content...
Please wait while we load your content...

