
Hybrid TiO2–acetylacetonate amorphous gel-derived material with stably adsorbed superoxide radical active in oxidative degradation of organic pollutants
Abstract
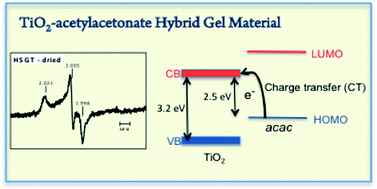
Hybrid sol–gel TiO2–acetylacetonate (HSGT) material has been synthesized via a sol–gel route. The as dried HSGT can be described as an amorphous polymeric network of titanium oxo-clusters on which surface part of Ti4+ ions are involved in strong complexation with acetylacetonate (acac) ligands. In this material a ligand-to-metal charge transfer (LMCT) is active giving light absorption in the visible region indicating a band gap of 2.5 eV, lower than crystalline TiO2. In the HSGT gel derived material the LMCT mechanism results much more active respect to ligands on crystalline nanoparticles. In presence of air, the acac ligands on the HSGT surface are able to generate and stabilize superoxide radical as resulting by EPR measurements. The ˙O2− radicals were stably adsorbed on the HSGT at room temperature for very long time. The presence of these free radicals makes HSGT material a useful catalyst in degradation of organic pollutants. Its catalytic activity has been tested in the oxidative degradation of phenanthrene resulting in a fast degradation rate in absence of any light irradiation.
 Please wait while we load your content...
Please wait while we load your content...

