
In situ mechanochemical synthesis of nitrones followed by 1,3-dipolar cycloaddition: a catalyst-free, “green” route to cis-fused chromano[4,3-c]isoxazoles†
Zigmee T. Bhutiaa,
Geethika P.a,
Anurag Malika,
Vikash Kumara,
Amrita Chatterjee*a,
Biswajit Gopal Royb and
Mainak Banerjee*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani-K. K. Birla, Goa Campus, Goa – 403 726, India. E-mail: mainak@goa.bits-pilani.ac.in; amrita@goa.bits-pilani.ac.in; Fax: +91-832-255-7031; Tel: +91-832-2580-347 Tel: +91-832-2580-320
bDepartment of Chemistry, Sikkim University, 6th Mile, Tadong, Gangtok, Sikkim-737102, India
First published on 13th November 2015
Abstract
An efficient and catalyst-free method for the synthesis of cis-fused chromano[4,3-c]isoxazoles via intramolecular 1,3-dipolar nitrone cycloaddition involving hand-grinding in a mortar-pestle has been developed. The mechanochemical agitation was sufficient for dehydrative nitrone formation by condensation of various O-allyl salicylaldehyde derivatives and alkyl/aryl hydroxylamines. The corresponding nitrones undergo intramolecular 1,3-dipolar cycloaddition leading to regioselective formation of cis-fused tetrahydrochromeno[4,3-c]isoxazole derivatives in high yields. The key features of this new method are cleaner reaction profiles, catalyst-free conditions, high yields, and short reaction times.
Introduction
Isoxazolidines,1 an important class of nitrogen containing five-membered heterocycles, is a ubiquitous structural motif of a wide spectrum of organic molecules of both natural origin and synthetic background, many of which are pharmaceutically important.2 1,3-Dipolar nitrone cycloaddition is the most facile way for the construction of these heterocycles as documented by different research groups.3 In particular, intramolecular nitrone–olefin cycloadditions are often employed to achieve structurally more complex bi- or tri-cyclic isoxazolidines of biological significance and also to synthesize key intermediates of several natural products.4 Notably, fused isoxazoles/isoxazolidines with chromano moiety are known to possess biomedical properties (Fig. 1) such as antidepressant, antipsychotic and antianxiolytic activities.5 Due to the labile nature of N–O bond chromanoisoxazoles are used as synthetic precursors for the construction of pharmaceutically important amino alcohols.6 Surprisingly, however, not many methods are available for the construction of these pharmacologically important heterocyclic systems.6a–c,7,8 Moreover, most of the existing methods are based on conventional synthetic protocols that use hazardous reagents, toxic solvents and/or relatively harsh reaction conditions and are facilitated by the presence of a catalyst.6a–c,7 Although few eco-friendly methods for chromano-isoxazoles are available,8 such a method can be turned more economical by avoiding use of catalysts, additional reagents and solvents.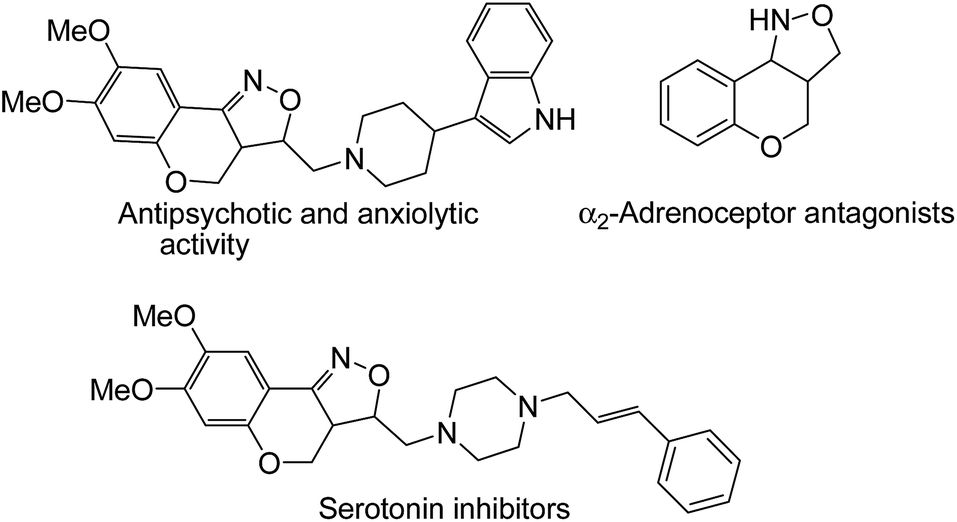 | ||
| Fig. 1 Chemical structures of few bioactive chromano[4,3-c]isoxazoles. | ||
Use of toxic chemicals and solvents for chemical transformations is a serious environmental concern for last few decades. At present, most of the chemical processes at an industrial scale use toxic organic solvents for various transformations which account for 80–90% of the waste generated in a typical pharmaceutical/fine chemical operational process.9 To counter this growing environmental problem, significant research efforts have been focused on solvent-free reactions,10 which are often associated with several other advantages such as faster reaction rates, lower energy consumption and easy separation giving rise to products in higher yields and with higher purities. One common technique employed in solvent-free reactions is mechanical grinding,11 which has gradually become a powerful tool in the paradigm of synthetic organic chemistry.12 In a typical mechanochemical process, reactions are initiated and progressed under frictional force provided either by grinding in a mortar-pestle or by milling in a ball-mill. Recently, mechanosynthesis by “ball milling” has emerged as effective technique for various organic transformations13,14 including aldol condensation,14a,b Michael additions,14c,d Knoevenagel condensation,14e Morita–Baylis–Hillman reactions,14f cross-coupling reactions,14h–k click reactions14l,m etc. On the other hand, manual grinding15 with a mortar and pestle is mostly limited to condensation reactions16 including Schiff's base formation,16a–c oxime formation,16d Knoevenagel condensation,16e with occasional exceptions.17 However, this a very useful method at laboratory scale due to simple and hazardless experimental set-up and is found to be equally effective for the construction of heterocyclic compounds of biological interest in last few years.18 In this purview, we envisaged, development of an environment friendly and a catalyst-free mechanochemical route to chromano-isoxazoles is a worthy pursuit. As per our current research focus of exploring the scope of hand-grinding techniques for organic transformation,18b herein, we report a one-pot process for nitrone formation followed by its intramolecular cycloaddition to afford a variety of cis-fused tetrahydrochromeno[4,3-c]isoxazole derivatives in high yields (Scheme 1).
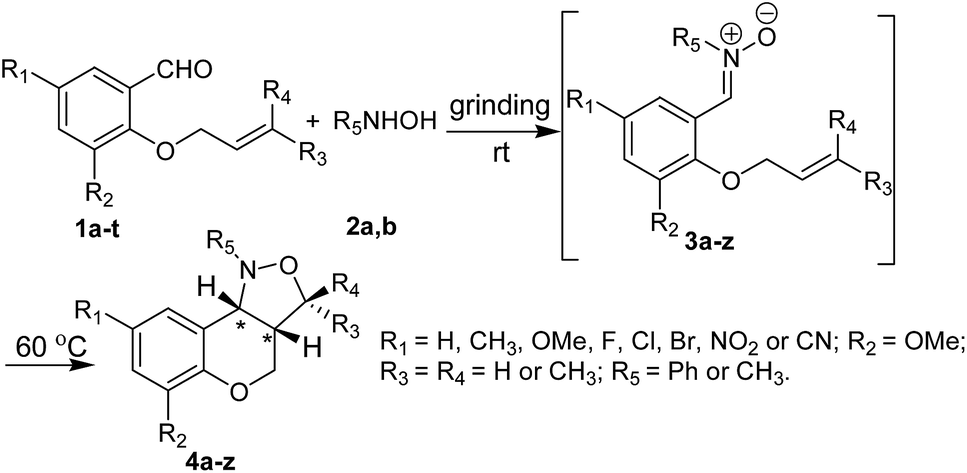 | ||
| Scheme 1 Mechanochemical route to cis-fused chromano[4,3-c]isoxazoles. | ||
Results and discussion
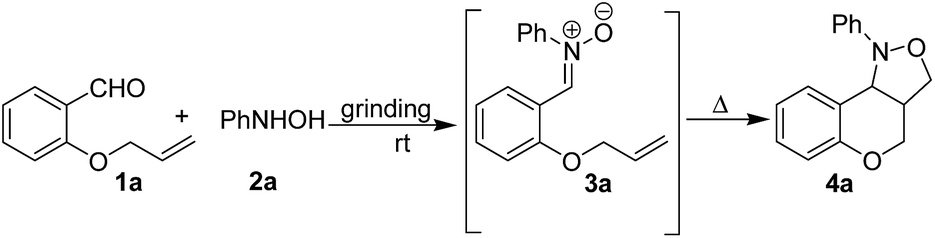
At first, we focused our attention on optimizing the reaction conditions. Thus, a model reaction was conducted between equimolar mixture of O-allyl salicylaldehyde (1a) and phenylhydroxylamine (2a) in an Agate mortar by manual grinding at room temperature to examine whether grinding under neat condition is useful or liquid assisted grinding (LAG)19 is more effective. The first reaction was conducted in neat condition. The reaction produced a viscous liquid after 5 min of gentle grinding. Although out of the two reactants N-phenylhydroxylamine is solid (mp 79–80 °C) the reaction mixture forms a melt phase presumably because of the transient heat generated during frictional force. The formation of nitrone was monitored by taking TLC after every 5 min. It was observed that grinding for 15 min in neat condition is sufficient for complete conversion of aldehyde (1a) and hydroxylamine (2a) to corresponding nitrone (3a). It is noteworthy to mention that rate of the reaction is dependent on the force applied for grinding the reaction mixture. As a matter of fact, fast and relentless grinding of the same reaction led to nitrone (3a) formation with about two third reduction in the reaction time (10 min). Since the nitrone formation is relatively fast even by gentle grinding the remaining reactions were carried out by gentle grinding only. However, intramolecular cycloaddition of nitrone to obtain chromano isoxazoles was bit slower. Only 20% of product (4a) was obtained even after 2 h of grinding of the intermediate nitrone (Table 1, entry 1). Cycloaddition was complete only after standing the mixture for 12 h at room temperature with intermittent grinding (Table 1, entry 1). However, gentle heating of the reaction mixture at 60 °C was helpful in almost 10 fold reduction of the reaction time. On the other hand, LAG effect was studied using three polar solvents viz. chloroform, ethanol and acetonitrile (0.5 mL per 1 mmol of substrate) in which all the starting materials, intermediates and products are freely soluble. Although nitrone formation was as fast as the neat reaction, there was practically no difference in the rate of conversion of nitrone (3a) into the chromano-isoxazole (4a) in each case of LAG. In addition, solvent got evaporated after sometimes and time to time addition of solvent (0.5 mL per 1 mmol of substrate each time) was required to continue LAG. Therefore, “neat grinding” was preferred over LAG for the synthesis of tetrahydrochromeno[4,3-c]isoxazole derivatives unless all the reactants are solid; in such cases, little amount of EtOH or 50% EtOH–H2O was used to form a paste which was ground further. It is noteworthy to mention that the reaction undergoes spontaneously without addition of any catalyst or additive making this a highly atom-efficient method for the synthesis of cis-fused chromano isoxazoles. In a separate study, the necessity of grinding for smooth formation of intermediate nitrone (3) was established by carrying out the same reaction in conventional ways (see ESI† for details). It was observed that nitrone formation is very sluggish in solution phase. At the same time, just mixing the reactants under neat condition without “grinding” is also not very effective. The nitrone formation was not complete even after 48 h. The formation of intermediate nitrone (3a) and the cyclized product (4a) was monitored by recording IR spectra of the reaction mixture at regular interval (see ESI† for details). It was observed that the characteristic stretching bands of starting materials like carbonyl of aromatic aldehyde at 1682 cm−1 and phenylhydroxylamine O–H and N–H bands at 3240 cm−1 and 3118 cm−1 almost disappeared after 10 min of hand grinding and a new peak at 1545 cm−1 (presumably, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching band of intermediate nitrone) appeared in the IR spectrum. The same band significantly diminished after gentle heating of the reaction mixture for 1.5 h indicating conversion of intermediate nitrone to chromano isoxazoles.
N stretching band of intermediate nitrone) appeared in the IR spectrum. The same band significantly diminished after gentle heating of the reaction mixture for 1.5 h indicating conversion of intermediate nitrone to chromano isoxazoles.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Timea (h) | (%) 3ab | (%) 4ab |
| a Reactions were ground for 10–120 min followed by heating.b Ratio of 3 and 4 was obtained from 1H NMR of reaction mixture. | |||||
| 1 | Neat | Rt | 0.25 | 100 | Nil |
| 2 | 70 | 20 | |||
| 12 | Nil | 86 | |||
| 60 | 1.5 | Nil | 84 | ||
| 2 | CHCl3 | Rt | 0.25 | 100 | Nil |
| 2 | 57 | 26 | |||
| 3 | EtOH | Rt | 0.17 | 100 | Nil |
| 2 | 70 | 18 | |||
| 4 | CH3CN | Rt | 0.34 | 100 | Nil |
| 2 | 64 | 25 | |||
To test the generality of this method, the phenolic –OH group of several salicylaldehyde derivatives were first alkylated with allyl group or prenyl group adopting reported procedure.20 Next, a series of O-allyl/prenyl derivatives of salicylaldehyde (1a–t) were ground with N-substituted hydroxylamines (2a,b) in an Agate mortar and pestle for several minutes to afford corresponding nitrones (Table 2). Once nitrone formation was complete (as revealed by TLC), the reaction mixture was heated on a sand bath at 60 °C for several hours to afford racemic cis-fused 1-aryl-1,3a,4,9b-tetrahydro-3H-chromano[4,3-c]isoxazoles (4a–z,aa) in excellent yields via in situ intramolecular cycloaddition in a stereoselective manner (Table 2). It is worthy to mention that all the intermediate nitrones (3) underwent complete conversion into chromano[4,3-c]isoxazoles (4) and the crude products were found to be sufficiently pure. Most of the crude products were purified by recrystallization from a mixture of ethyl acetate and petroleum ether. Only few of the final products, which were obtained as viscous liquid, were purified by passing them through a short bed of silica gel. It is noteworthy to mention that N-methylhydroxylamine (2b) was generated from corresponding hydrochloride salt in situ by addition of sodium carbonate to the reaction mixture. For these reactions few drops of 50% EtOH–water was added at the beginning and the resulting paste was ground thoroughly with portionwise addition of Na2CO3. It was observed that the nitrone formation was much faster in the presence of little amount of solvent than at neat condition. Most likely, EtOH–water mixture dissolves a part of CH3NHOH·HCl and Na2CO3 making release of N-methylhydroxylamine easy. The products derived from N-methylhydroxylamine (2b) were taken in ethyl acetate and washed with water to remove sodium carbonate if any and then purified either by crystallization or by column chromatography. All the chromano[4,3-c]isoxazoles were characterized by 1H NMR, 13C NMR, ESI-MS and CHN analysis. The spectra of known compounds were in well agreement with the reported values.7b,c,8b Notably, Yadav et al.7b and we8b separately demonstrated that 1,3-dipolar cycloaddition of nitrones derived from O-allyl salicylaldehyde derivatives preferably form chromano[4,3-c]isoxazoles with cis-stereochemistry at the junction of six- and five-membered rings. The expected cis stereochemistry was verified by comparing the coupling constant (JH3a–H9b) of ring junction protons of the compounds synthesized using the current method with chromano[4,3-c]isoxazoles that are previously reported by our group.8b A relatively small coupling constant (see Table S2 of ESI†) between ring junction protons of all the chromano isoxazoles clearly indicate that the five- and six-membered rings adopt a cis-fused twisted structure.7b,8b
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Salicylaldehyde derivatives | R5 | Timea (min) nitrone | Timeb (h) product | Product | % Yieldc | Ref. |
| a Reactions were ground for 10–40 minutes for intermediate nitrone formation.b The reaction mixtures were heated on a sand bath for several hours.c All yields refer to isolated product, characterised by 1H-NMR,13C-NMR, ESI-MS. | |||||||
| 1 | 1a: R1 = R2 = R3 = R4 = H | Ph | 15 | 1.5 | 4a | 86 | 8b |
| 2 | 1b: R1 = R2 = H, R3 = R4 = CH3 | Ph | 15 | 3.0 | 4b | 79 | 7b and 8b |
| 3 | 1a: R1 = R2 = R3 = R4 = H | CH3 | 20 | 3.0 | 4c | 71 | 7c |
| 4 | 1b: R1 = R2 = H, R3 = R4 = CH3 | CH3 | 20 | 4.0 | 4d | 70 | — |
| 5 | 1c: R1 = Br, R2 = R3 = R4 = H | Ph | 15 | 2.0 | 4e | 80 | 8b |
| 6 | 1d: R1 = Br, R2 = H, R3 = R4 = CH3 | Ph | 15 | 2.5 | 4f | 87 | 7b and 8b |
| 7 | 1c: R1 = Br, R2 = R2 = R3 = H | CH3 | 20 | 3.0 | 4g | 68 | — |
| 8 | 1e: R1 = OMe, R2 = R3 = R4 = H | Ph | 30 | 4.0 | 4h | 88 | — |
| 9 | 1f: R1 = OMe, R2 = H, R3 = R4 = CH3 | Ph | 30 | 4.0 | 4i | 90 | — |
| 10 | 1g: R1 = H, R2 = OMe, R3 = R4 = H | Ph | 30 | 3.0 | 4j | 89 | 8b |
| 11 | 1h: R1 = H, R2 = OMe, R3 = R4 = CH3 | Ph | 30 | 4.5 | 4k | 91 | 7b and 8b |
| 12 | 1g: R1 = H, R2 = OMe, R3 = R4 = H | CH3 | 40 | 2.5 | 4l | 70 | — |
| 13 | 1i: R1 = Cl, R2 = R3 = R4 = H | Ph | 10 | 2.5 | 4m | 83 | — |
| 14 | 1j: R1 = Cl, R2 = H, R3 = R4 = CH3 | Ph | 20 | 4.0 | 4n | 88 | — |
| 15 | 1k: R1 = CH3, R2 = R3 = R4 = H | Ph | 15 | 2.0 | 4o | 82 | — |
| 16 | 1l: R1 = CH3, R2 = H, R3 = R4 = CH3 | Ph | 15 | 4.0 | 4p | 87 | — |
| 17 | 1m: R1 = F, R2 = R3 = R4 = H | Ph | 10 | 1.5 | 4q | 84 | — |
| 18 | 1n: R1 = F, R2 = H, R3 = R4 = CH3 | Ph | 10 | 3.0 | 4r | 87 | — |
| 19 | 1m: R1 = F, R2 = R3 = R4 = H | CH3 | 15 | 3.0 | 4s | 69 | — |
| 20 | 1o: R1 = NO2, R2 = R3 = R4 = H | Ph | 10 | 1.0 | 4t | 84 | 8b |
| 21 | 1q: R1 = CN, R2 = R3 = R4 = H | Ph | 10 | 1.0 | 4u | 83 | — |
| 22 | 1r: R1 = CN, R2 = H, R3 = R4 = CH3 | Ph | 15 | 1.5 | 4v | 85 | — |
| 23 | 1q: R1 = CN, R2 = R3 = R4 = H | CH3 | 20 | 3.0 | 4w | 63 | — |
| 24 | 1s: 2-(Allyloxy)naphthalene-1-carbaldehyde | Ph | 20 | 1.0 | 4x | 91 | — |
| 25 | 1t: 2-(3-Methylbut-2-enyloxy)naphthalene-1-carbaldehyde | Ph | 30 | 1.0 | 4y | 94 | — |
| 26 | 1s: 2-(Allyloxy)naphthalene-1-carbaldehyde | CH3 | 30 | 2.0 | 4z | 79 | — |
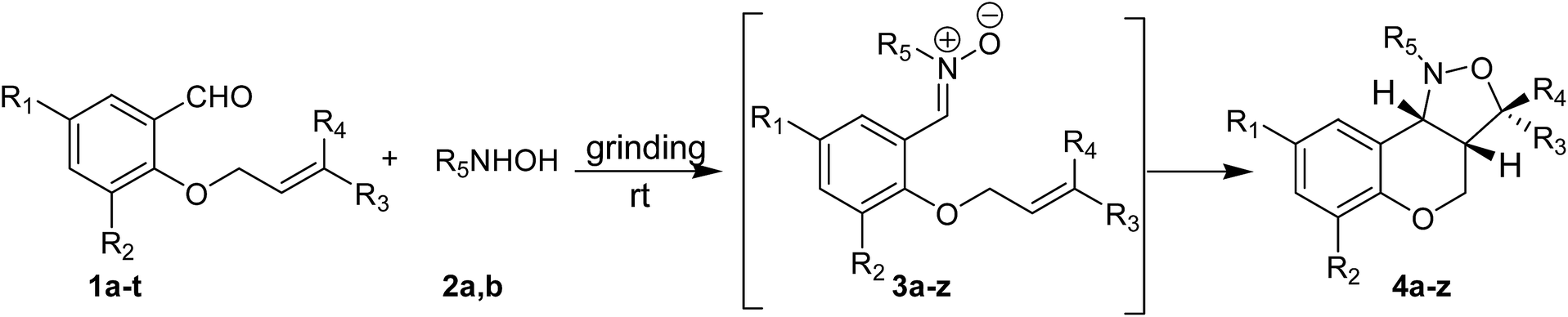
In general, the method worked well with both aliphatic and aromatic hydroxylamines and had been applied to a variety of O-allyl salicylaldehydes with same efficacy. Noticeably, substituents in the aromatic ring of the O-allyl salicylaldehyde derivatives did not pose any significant effect on the yield of chromano[4,3-c]isoxazoles. However, yields of chromano[4,3-c]isoxazoles derived from N-methylhydroxylamine (2b) (Table 2, entry 7, 19, 23 etc.) were slightly less than that of N-phenylhydroxylamine (2a) (Table 2, entry 5, 17, 21 etc.). It was also observed that the nitrone formation for a particular O-allyl salicylaldehyde derivative was little faster with N-phenylhydroxylamine (2a) (Table 2, entry 1, 2, 5, 18 etc.) as compared to N-methylhydroxylamine (2b) (Table 2, entry 3, 4, 7, 19 etc.). Moreover, the intramolecular cycloaddition was generally faster for nitrones derived from N-phenylhydroxylamine (2a) (Table 2, entry 1, 5, 20, 21 etc.) than that of N-methylhydroxylamine (2b) (Table 2, entry 3, 7, 19 etc.). Presumably, the electron donation ability of the methyl group makes the 1,3-dipolarophile less reactive, whereas, phenyl group acts as an electron pulling unit to make nitrone more reactive. Again, doubly substituted allyl moiety (i.e. prenyl group) although did not influence the yield of 4 but slowed down the reaction due to steric reason (Table 2, entry 2, 4, 6, 18 etc.).
Conclusion
In conclusion, we have developed a catalyst-free method for the synthesis of cis-fused chromano[4,3-c]isoxazoles via intramolecular 1,3-dipolar nitrone cycloaddition reaction involving hand-grinding in mortar-pestle. A series of O-allyl salicylaldehyde derivatives were successfully condensed with alkyl/aryl hydroxylamines to produce corresponding chromano[4,3-c]isoxazoles in high yields. Most of the reactions were conducted under solvent-free condition and in few cases, minimum volume of ethanol–water was used for proper mixing of reactants. The approach is “greener” and more advantageous over existing methods because of drastic reduction in the use of organic solvents accompanied with clean reaction profile, high yields, and short reaction times.Experimental
General information
All the reagents were procured from commercial sources and were used without further purification. All solvents were obtained from local suppliers and were of research grade. 1H NMR and 13C NMR spectra were recorded on Bruker Avance (300 or 400 MHz, respectively) with TMS or solvent peak as internal standard. The chemical shifts are reported in parts per million (ppm) units. Mass spectra were recorded on Agilent 6220 Accurate-Mass TOF LC-MS using ESI as the ion source. IR spectra were recorded in KBr pellets with IR Affinity 1, Shimadzu. CHN data were recorded using Vario MICRO elementar CHNS analyzer. Melting points of the compounds were determined using Melting Point Apparatus, Bio Techniques, India. The reactions were monitored by thin layer chromatography (TLC) carried out on 0.25 mm silica gel on aluminium plates (60F-254) using UV light (254 or 365 nm). Column chromatography was performed on silica gel (60–120 mesh, Merck).General procedure for chromano[4,3-c]isoxazoles: synthesis of 4h
2-(Allyloxy)-5-methoxybenzaldehyde (1e, 192 mg, 1 mmol) and phenylhydroxylamine (2a, 115 mg, 1.05 mmol) was taken in a Agate mortar and the mixture was ground thoroughly by a pestle for 30 min. The complete conversion of starting materials to nitrone (3h) was monitored by TLC. Next, the mortar was placed in a sand bath and the reaction mixture was heated at 60 °C for 4 h. The crude product was recrystallized from 20% EtOAc in petroleum ether to afford corresponding chromeno[4,3-c]isoxazole, 4h in pure form (248 mg, 88%).Selected spectral data of new entries
Acknowledgements
M. B. thanks CSIR (India) (project No. 02(0075)/12/EMR-II) for financial support. Z. T. B. is thankful to DST India for INSPIRE fellowship and V. K. is indebted to CSIR, India for SRFship.Notes and references
- P. Grunanger and P. Vita-Finzi, Isoxazoles, Wiley, New York, 1991 Search PubMed.
- (a) R. Romeo, M. Navarra, S. V. Giofrè, C. Carnovale, S. Cirmi, G. Lanza and M. A. Chiacchio, Bioorg. Med. Chem., 2014, 22, 3379 CrossRef CAS PubMed; (b) P. P. Shao, F. Ye, A. E. Weber, X. Li, K. A. Lyons, W. H. Parsons, M. L. Garcia, B. T. Priest, M. M. Smith, J. P. Felix, B. S. Williams, G. J. Kaczorowski, E. McGowan, C. Abbadie, W. J. Martin, D. R. McMasters and Y. D. Gao, Bioorg. Med. Chem. Lett., 2009, 19, 5334 CrossRef CAS PubMed; (c) A. Piperno, S. V. Giofré, D. Iannazzo, R. Romeo, G. Romeo, U. Chiacchio, A. Rescifina, D. Iannazzo, A. Piperno, A. Rescifina, R. Romeo, M. Saglimbeni, T. Sciortino, V. Valveri and A. G. Piotrowska, J. Org. Chem., 2010, 75, 2798 CrossRef CAS PubMed; (d) U. Chiacchio, E. Balestrieri, B. Macchi, D. Mastino and G. Romeo, J. Med. Chem., 2005, 48, 1389 CrossRef CAS PubMed; (e) A. Rescifina, M. A. Chiacchio, A. Corsaro, E. D. Clercq, D. Iannazzo, A. Mastino, A. Piperno, G. Romeo, R. Romeo and V. Valveri, J. Med. Chem., 2006, 49, 709 CrossRef CAS PubMed; (f) U. Chiacchio, D. Iannazzo, A. Piperno, R. Romeo, G. Romeo, A. Rescifina and M. Saglimbeni, Bioorg. Med. Chem., 2006, 14, 955 CrossRef CAS PubMed; (g) M. P. Sadashiva, H. Mallesha, N. A. Hitesh and K. S. Rangappa, Bioorg. Med. Chem., 2004, 12, 6389 CrossRef CAS PubMed.
- (a) T. Mita, N. Ohtsuki, T. Ikeno and T. Yamada, Org. Lett., 2002, 4, 2457 CrossRef CAS PubMed; (b) S. Saubern, J. M. Macdonald, J. H. Ryan, R. C. J. Woodgate, T. S. Louie, M. J. Fuchter, J. M. White and A. B. Holmes, Tetrahedron, 2010, 66, 2761 CrossRef CAS; (c) K. Rück-Braun, T. H. E. Freysoldt and F. Wierschem, Chem. Soc. Rev., 2005, 34, 507 RSC; (d) C. Lu, A. V. Dubrovskiy and R. C. Larock, J. Org. Chem., 2012, 77, 2279 CrossRef CAS PubMed; (e) K. Moriyama, Y. Izumisawa and H. Togo, J. Org. Chem., 2011, 76, 7249 CrossRef CAS PubMed; (f) E. Falkowska, M. Y. Laurent, V. Tognetti, L. Joubert, P. Jubault, J.-P. Bouillon and X. Pannecoucke, Tetrahedron, 2015, 71, 8067 CrossRef CAS.
- (a) M. Yamaguchi, A. Matsuda and S. Ichikawa, Org. Biomol. Chem., 2015, 13, 1187 RSC; (b) A. Aguiar, A. Leite, A. M. N. Silva, A. C. Tomé, L. Cunha-Silva, B. D. Castro, M. Rangel and A. M. G. Silva, Org. Biomol. Chem., 2015, 13, 7131 RSC; (c) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247 CrossRef CAS; (d) J. H. Jeong and S. M. Weinreb, Org. Lett., 2006, 8, 2309 CrossRef CAS PubMed; (e) O. Tamura, N. Iyama and H. Ishibashi, J. Org. Chem., 2004, 69, 1475 CrossRef CAS PubMed; (f) D. D. Dhavale, S. M. Jachak, N. P. Karche and C. Trombini, Tetrahedron, 2004, 60, 3009 CrossRef CAS; (g) J. D. White and J. D. Hansen, J. Am. Chem. Soc., 2002, 124, 4950 CrossRef CAS PubMed.
- (a) N. F. L. Machado and M. P. M. Marques, Curr. Bioact. Compd., 2010, 6, 76 CrossRef CAS; (b) S. Singh, A. Chopra, G. Singh, A. K. Saxena, M. Paul and S. Ishar, J. Pharmacol. Res., 2013, 7, 337 CrossRef CAS; (c) J. I. Andres-Gil, J. M. Bartolome-Nebreda, M. J. Alcazar-Vaca, M. D. l. M. Gracia-Martin, A. A. H. P. Megens, US Pat., 113988, 2008; (d) J. I. Andrés, J. Alcázar, J. M. Alonso, R. M. Alvarez, M. H. Bakker, I. Biesmans, J. M. Cid, A. I. D. Lucas, W. Drinkenburg, J. Fernández, L. M. Font, L. Iturrino, X. Langlois, I. Lenaerts, S. Martínez, A. A. Megens, J. Pastor, S. Pullan and T. Steckler, Bioorg. Med. Chem., 2007, 15, 3649 CrossRef PubMed; (e) J. Pastor, J. Alcázar, R. M. Alvarez, J. I. Andrés, J. M. Cid, A. I. D. Lucas, A. Díaz, J. Fernández, L. M. Font, L. Iturrino, C. Lafuente, S. Martínez, M. H. Bakker, I. Biesmans, L. I. Heylen and A. A. Megens, Bioorg. Med. Chem. Lett., 2004, 14, 2917 CrossRef CAS PubMed; (f) J. I. Andrés, J. Alcázar, J. M. Alonso, R. M. Alvarez, J. M. Cid, A. I. D. Lucas, J. Fernández, S. Martínez, C. Nieto, J. Pastor, M. H. Bakker, I. Biesmans, L. I. Heylen and A. A. Megens, Bioorg. Med. Chem. Lett., 2003, 13, 2719 CrossRef.
- (a) Q. Zhao, F. Han and D. L. Romero, J. Org. Chem., 2002, 67, 3317 CrossRef CAS PubMed; (b) G. Broggini, L. Bruche, E. Cappelletti and G. Zecchi, J. Chem. Res., Synop., 1997, 36 RSC; (c) G. Broggini, F. Folcio, N. Sardone, M. Sonzogni and G. Zecchi, Tetrahedron: Asymmetry, 1996, 7, 797 CrossRef CAS; (d) M. Frederickson, Tetrahedron, 1997, 53, 403 CrossRef CAS.
- (a) M. Bakthadoss and G. Murugan, Eur. J. Org. Chem., 2010, 2010, 5825 CrossRef; (b) J. S. Yadav, B. V. S. Reddy, D. Narsimhaswamy, K. Narsimulub and A. C. Kunwar, Tetrahedron Lett., 2003, 44, 3697 CrossRef CAS; (c) C. Baldoli, P. Del Buttero, E. Licandro, S. Maiorana and A. Papagni, Tetrahedron: Asymmetry, 1995, 6, 1711 CrossRef CAS; (d) T. Aftab, R. Grigg, M. Ladlow, V. Sridharan and M. Thornton-Pett, Chem. Commun., 2002, 1754 RSC; (e) A. Abiko, Chem. Lett., 1995, 357 CrossRef CAS.
- (a) M. J. Raihan, V. Kavala, C.-W. Kuo, B. R. Raju and C.-F. Yao, Green Chem., 2010, 12, 1090 RSC; (b) A. Chatterjee, S. K. Hota, M. Banerjee and P. K. Bhattacharya, Tetrahedron Lett., 2010, 51, 6700 CrossRef CAS.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- (a) K. Tanaka and F. Toda, Solvent-free Organic Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) A. Kumar and S. Sharma, Green Chem., 2011, 13, 2017 RSC; (c) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140 CrossRef CAS PubMed; (d) M. S. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547 RSC; (e) S. Yan, Y. Chen, L. Liu, N. He and J. Lin, Green Chem., 2010, 12, 2043 RSC; (f) M. S. Singh, G. C. Nandi and S. Samai, Green Chem., 2012, 14, 447 RSC.
- (a) K. D. M. Harris, Nat. Chem., 2013, 5, 12 CrossRef CAS PubMed; (b) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC.
- (a) B. C. Ranu and A. Stolle, Ball Milling Towards Green Synthesis: Applications, Projects, Challenges, Royal Society of Chemistry, Cambridge, 2014 Search PubMed; (b) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC.
- For selected recent examples of organic synthesis in ball-mills, see: (a) J. G. Hernandez and E. Juaristi, J. Org. Chem., 2011, 76, 1464 CrossRef CAS PubMed; (b) J. G. Hernández, V. García-López and E. Juaristi, Tetrahedron, 2012, 68, 92 CrossRef; (c) Y.-F. Wang, R.-X. Chen, K. Wang, B.-B. Zhang, Z.-B. Lib and D.-Q. Xu, Green Chem., 2012, 14, 893 RSC; (d) M. Jörres, S. Mersmann, G. Raabe and C. Bolm, Green Chem., 2013, 15, 612 RSC; (e) R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 873 RSC; (f) J. Mack and M. Shumba, Green Chem., 2007, 9, 328 RSC; (g) D. C. Waddell and J. Mack, Green Chem., 2009, 11, 79 RSC; (h) F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894 RSC; (i) G. Cravotto, D. Garella, S. Tagliapietra, A. Stolle, S. Schüsler, S. E. S. Leonhardt and B. Ondruschka, New J. Chem., 2012, 36, 1304 RSC; (j) D. A. Fulmer, W. C. Shearouse, S. T. Medonza and J. Mack, Green Chem., 2009, 11, 1821 RSC; (k) R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985 RSC; (l) R. Thorwirth, A. Stolle, B. Ondruschka, A. Wild and U. S. Schubert, Chem. Commun., 2011, 47, 4370 RSC; (m) T. L. Cook, J. A. Walker and J. Mack, Green Chem., 2013, 15, 617 RSC; (n) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Commun., 2013, 49, 591 RSC; (o) W. Su, J. Yu, Z. Li and Z. Jiang, J. Org. Chem., 2011, 76, 9144 CrossRef CAS PubMed; (p) V. Štrukil, B. Bartolec, T. Portada, I. Ðilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100 RSC; (q) J. G. Hernández and E. Juaristi, J. Org. Chem., 2010, 75, 7107 CrossRef PubMed; (r) D. Tan, V. Štrukil, C. Mottillo and T. Friščić, Chem. Commun., 2014, 50, 5248 RSC; (s) Y. Fang, N. Salamé, S. Woo, D. S. Bohle, T. Friščić and L. A. Cuccia, CrystEngComm, 2014, 16, 7180 RSC; (t) K. Crossey, R. N. Cunningham, P. Redpath and M. E. Migaud, RSC Adv., 2015, 5, 58116 RSC; (u) I. Dokli and M. Gredičak, Eur. J. Org. Chem., 2015, 2015, 2727 CrossRef CAS; (v) P. F. M. Oliveira, M. Baron, A. Chamayou, C. André-Barrès, B. Guidetti and M. Baltas, RSC Adv., 2014, 4, 56736 RSC; (w) T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Eur.–J., 2015, 21, 12787 CrossRef PubMed; (x) T. K. Achar, S. Maiti and P. Mal, RSC Adv., 2014, 4, 12834 RSC.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS PubMed; (b) F. Toda, Acc. Chem. Res., 1995, 28, 480 CrossRef CAS.
- (a) O. Dolotko, J. W. Wiench, K. W. Dennis, V. K. Pecharsky and V. P. Balema, New J. Chem., 2010, 34, 25 RSC; (b) D. Cinčić, I. Brekalo and B. Kaitner, Chem. Commun., 2012, 48, 11683 RSC; (c) D. Cinčić, I. Brekalo and B. Kaitner, Cryst. Growth Des., 2012, 12, 44 CrossRef; (d) C. B. Aakeröy and A. S. Sinha, RSC Adv., 2013, 3, 8168 RSC; (e) S. I. Bhat, A. R. Choudhury and D. R. Trivedi, RSC Adv., 2012, 2, 10556 RSC; (f) L. P. Jameson and S. V. Dzyuba, Beilstein J. Org. Chem., 2013, 9, 786 CrossRef CAS PubMed; (g) N. M. Rateb and H. F. Zohdi, Synth. Commun., 2009, 39, 2789 CrossRef CAS.
- (a) V. Štrukil, M. D. Igrc, L. Fábián, M. Eckert-Maksić, S. L. Childs, D. G. Reid, M. J. Duer, I. Halasz, C. Mottilloe and T. Friščić, Green Chem., 2012, 14, 2462 RSC; (b) V. Štrukil, M. D. Igrc, M. Eckert-Maksić and T. Friščić, Chem. Eur.–J., 2012, 18, 8464 CrossRef PubMed; (c) I. Huskić, I. Halasz, T. Friščić and H. Vančik, Green Chem., 2012, 14, 1597 RSC.
- (a) S. Majumdar, M. Chakraborty, N. Pramanik and D. K. Maiti, RSC Adv., 2015, 5, 51012 RSC; (b) M. Banerjee, A. Chatterjee, V. Kumar, Z. T. Bhutia, D. G. Khandare, M. S. Majik and B. G. Roy, RSC Adv., 2014, 4, 39606 RSC; (c) A. Khaskel, P. Gogoi, P. Barman and B. Bandyopadhyay, RSC Adv., 2014, 4, 35559 RSC; (d) H. Shy, P. Mackin, A. S. Orvieto, D. Gharbharan, G. R. Peterson, N. Bampos and T. D. Hamilton, Faraday Discuss., 2014, 170, 59 RSC; (e) G. Brahmachari and S. Das, RSC Adv., 2014, 4, 7380 RSC; (f) G. Shukla, G. K. Verma, A. Nagaraju, R. K. Verma, K. Raghuvanshi and M. S. Singh, RSC Adv., 2013, 3, 13811 RSC.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334 RSC.
- R. Rohlmann, C.-G. Daniliuc and O. G. Manchenõ, Chem. Commun., 2013, 49, 11665 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, IR studies, selected spectra of compounds, etc. See DOI: 10.1039/c5ra21044e |
| This journal is © The Royal Society of Chemistry 2015 |