
Selective recovery of rare earth elements using chelating ligands grafted on mesoporous surfaces†
Abstract
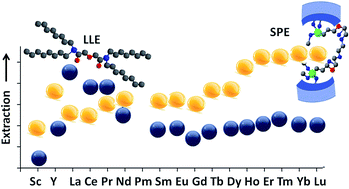
Nowadays, rare earth elements (REEs) and their compounds are critical for the rapidly growing advanced technology sectors and clean energy demands. However, their separation and purification still remain challenging. Among different extracting agents used for REE separation, the diglycolamide (DGA)-based materials have attracted increasing attention as one of the most effective extracting agents. In this contribution, a series of new and element-selective sorbents were generated through derivatisation of the diglycolamide ligand (DGA), grafted to mesoporous silica and tested for the separation of rare earth elements. It is shown that, by tuning the ligand bite angle and its environment, it is possible to improve the selectivity towards specific rare earth elements.
 Please wait while we load your content...
Please wait while we load your content...

