
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted telescoped cross metathesis-ring closing aza-Michael reaction sequence: step-economical access to nicotine–lobeline hybrid analogues†
E. Drège,
J. Oko,
P.-E. Venot,
N. Gigant and
D. Joseph*
Université Paris-Sud, BioCIS, Equipe de Chimie des Substances Naturelles, Université Paris-Saclay 5, rue Jean-Baptiste Clément, F-92296 Châtenay-Malabry, France. E-mail: delphine.joseph@u-psud.fr
First published on 6th November 2015
Abstract
A series of 2,5-disubstituted pyrrolidines was synthesized through an efficient telescoped cross-metathesis/cyclizing aza-Michael addition involving N-heteroaromatic olefinic derivatives. This synthetic route was applied to the preparation of original nicotine–lobeline, nicotine–pelletierine and lobeline–nicotine–epibatidine hybrids.
Introduction
Nicotinic acetylcholine receptors (nAChRs) belong to the family of pentameric ligand-gated channels. As they play a significant role in cognitive and sensory gating processes, nAChRs comprise potentially therapeutic targets in manifold brain disorders.1 One of the major challenges in drug discovery targeting nAChRs, is to develop compounds that can selectively bind one receptor subtype. If fragment-based approach to find out selective receptor ligands is nowadays widely established in drug discovery and chemical biology, the application of this concept on nAChRs is delayed due to the lack of readily available structural information and sensitive biophysical screening methods. Only one fundamental example of a fragment merging optimization of ligand has been described for the acetylcholine binding protein, a model protein for the extracellular α7 nAChR-subtype domain.2 Therefore, the concept of molecular hybridization continues to be an alternative answer to find out new nAChRs ligands. More potent and selective ligands have been developed by combining the pharmacophores of natural alkaloids that exhibit a pronounced nAChR pharmacological activity such as nicotine (1), epibatidine (2), anatoxine (3) and lobeline (4) (Fig. 1).3 | ||
| Fig. 1 Naturally occurring nAChR ligands and nicotine–lobeline hybrid analogues 5. | ||
A part of our research program aims at shaping new step-economical synthetic processes of new Lobelia alkaloids analogues as ligands of nAChR-subtypes.4 In the context of targeting α4β2-subtype, the synthesis of hybrid molecules appeared to us as an interesting challenge and we decided to investigate the preparation of original lobeline–natural nAChR ligands chimeric analogues 5 by connecting relevant pharmacophores (Fig. 1).
A key advantage of the concept of molecular hybridization is its capacity to create highly chemically diverse molecules with a high degree of congenital resemblance, an essential criterion for relevant structure–activity relationship studies. We thus made an effort at designing a diastereoselective synthetic pathway that could rapidly reach structural and functional diversity. The shortness and the flexibility of this synthetic strategy will be insured by a cross metathesis-ring closing aza-Michael (CM-RCAM) sequence (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic strategy for the preparation of hybrids 5. | ||
One of the challenges of our approach lies in the cross-metathesis of tethering N-heteroaromatic-containing olefinic substrates 6 with sensitive electron-poor olefinic coupling partners 7. Retrosynthetic analysis suggests that the transformation of the hydroxyl group of 8 into an amine function could easily lead to the formation of the key CM-RCAM precursors 6. It was further expected that the synthesis of the alcohol 8 would be secured by the condensation and 3-butenylmagnesium bromide with 2-substituted-5-carboxaldehydes 10.
We present herein a short and efficient access to nicotine–natural nAChR ligand hybrids for which each synthesis step offers both diversity and flexibility with three distinct sites of modulation (R1, R2 and R3) and involves simple and commercially available precursors (7, 9 and 10).
Results & discussion
Our synthetic efforts are depicted in Scheme 2 and started with the efficient preparation of the pent-4-en-1-ol 8a and 8b. The olefinic Grignard reagent was easily prepared starting from 4-bromo-1-butene in the presence of magnesium before reacting with the commercially available pyridine-3-carboxaldehyde 10a or 6-chloropyridine-3-carboxaldehyde 10b.5 The hydroxyl function of the pyridinic pentenols 8 was then efficiently transformed in a leaving group by mesylation under standard conditions. The mesylates 11a and 11b were isolated without further purification in 96% and 92% yields, respectively. Our initial attempts to synthesize the required amine moiety were envisaged via a two-step sequence including the introduction of an azido group followed by its reduction into primary amine. Treatment of the mesylated derivative 11a with sodium azide led to the desired azide 12a in a 80% yield. Unfortunately, the Staudinger reduction of 12a under usual conditions failed, giving a complex mixture of products. In the same way, other methods for converting an azido group into an amine function using propane-1,3-dithiol,6 or indium metal and ammonium chloride7 gave a mixture of inseparable polar products. | ||
Scheme 2 Reagents and conditions for the synthesis of 4-penten-1-amines 6a and 6b: (a) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2MgBr, THF, −20 °C to rt; (b) MsCl, Et3N, CH2Cl2; (c) MeNH2, DMF, H2O, or MeNH2 (2 M, in THF) 50 °C; (d) NaN3, DMF; (e) Boc2O, Et3N, CH2Cl2. CHCH2CH2MgBr, THF, −20 °C to rt; (b) MsCl, Et3N, CH2Cl2; (c) MeNH2, DMF, H2O, or MeNH2 (2 M, in THF) 50 °C; (d) NaN3, DMF; (e) Boc2O, Et3N, CH2Cl2. | ||
As the transformation of the azide function appeared to be quite problematic, an alternative synthetic pathway was investigated. In this way, we evidenced that the mesylate 11a easily underwent nucleophilic substitution (SN2) in the presence of aqueous methylamine in DMF at 60 °C, affording the pentenamine 14a in a satisfying 60% yield. The yield was increased to 75% by using a 2 M THF solution of methylamine rendering the purification step simpler as well. These amination conditions were also successfully applied to 11b delivering 14b in a good 70% yield. Nonetheless, a longer reaction time was necessary to reach full conversion: four days were needed for 14b instead of twelve hours in the case of 14a suggesting a deactivating effect of the chlorine atom on the nucleophilic substitution. Interestingly, it must be pointed out that these N-methyl-pent-4-en-1-amines 14 did not require any further purification, before being engaged in the following step, highlighting the synthetic process cleanliness. At this stage of our synthesis, the protection of the methylamine group of 14 by an electron-withdrawing group was envisaged to avoid potential deactivating coordination between the amine function by the ruthenium-based catalyst during the olefin cross-metathesis (CM). N-Methylamines 14 were thus treated with Boc2O providing the advanced key intermediates 6a and 6b in high yields (90% and 75%, respectively).
Only few examples of tandem CM/aza-Michael process have already been reported in the literature.4c,8 The bibliography becomes particularly poor for substrates bearing a strong Lewis base such as a free amine or a pyridine that may dramatically deactivate the catalyst.9 To the best of our knowledge, the reactivity of these potentially metal-coordinating substrates has never been described in the presence of electronically biased olefins, such as α,β-unsaturated ketones. Following our recent results in this field,4c we decided to screen the efficiency of five commercially available Ru complexes (Fig. 2). These pre-catalysts were selected based on their catalytic features: three standard metathesis complexes such as the Grubbs 2nd generation I (G-II),10a the Grubbs–Hoveyda 2nd generation II (GH-II)10b and the indenylidene-based complex III (M2);10c and two well-defined fast-initiation Hoveyda-type complexes bearing either a SIMes (IV) or a SIPr (V) NHC unit.11
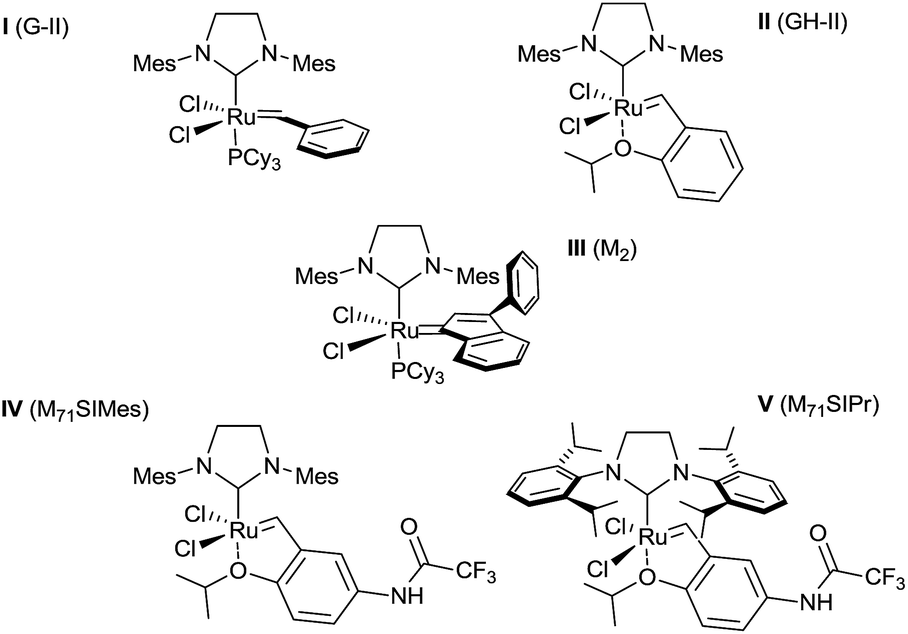 | ||
| Fig. 2 Screened ruthenium-based metathesis complexes. | ||
We first evaluated the feasibility of this reaction by submitting the pyridinic olefinic derivatives 14 and 6 with methyl acrylate 7a or ethyl acrylate 7b. As earlier described by the group of Cossy on pyridinic homoallylic alcohols,9a the CM involving the pyridinic olefinic amine 14a in the presence of the Grubbs catalysts I or II (10 mol%) in refluxing dichloromethane for 24 h only permitted the recovery of the starting material. These results were in coherence with the known poisonous character of both the amine and the pyridine substituents which deactivate the ruthenium catalyst by coordinating the metal centre.9b The study was thus pursued using the pyridines 6a and 6b and our results are summarized in Table 1. In this way, starting from the N-Boc protected derivative 6a, the desired cross-coupled product 15aa was formed in an encouraging 55% yield in the presence of the 2nd generation Grubbs catalyst I under the same aforementioned reaction conditions (Table 1, entry 1). The use of GH-II catalyst (II) provided the CM product 15aa in an enhanced 60% yield even with a lower 5 mol% catalyst loading (Table 1, entries 2 and 3). Moreover, the reaction time was dramatically reduced from 24 h to 45 min or 60 min when microwave heating was employed (Table 1, entries 1–3). Interestingly, yields were improved up to 70% by using more hindered catalysts such as IV or V with ethyl acrylate 7b as olefinic partner (Table 1, entries 4–6).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Cat. (mol%) | Product | Thermal yieldb (%) | μwaves yieldb (%) (time) |
| a Conditions: 1 equiv. 6a or 6b, 1.3 equiv. 7a–7d, CH2Cl2 (0.5 M), reflux, 24 h or, 100 °C μwaves (200 W).b Isolated yield.c The reactions were only performed under microwave irradiation.d Starting material. | ||||||
| 1 | H | OMe | I (10) | 15aa | 55 | 65 (1 h) |
| 2 | H | OMe | II (10) | 15aa | 60 | 70 (1 h) |
| 3 | H | OMe | II (5) | 15aa | 60 | 70 (45 min) |
| 4c | H | OEt | III (10) | 15ab | — | 30 (3 h) |
| 5c | H | OEt | IV (5) | 15ab | — | 70 (45 min) |
| 6c | H | OEt | V (5) | 15ab | — | 70 (45 min) |
| 7 | H | Me | II (5) | 15ac | 50 | 70 (1 h) |
| 8 | H | Ph | II (5) | 15ad | SMd | 50 (3 h) |
| 9c | H | Ph | III (5) | 15ad | — | 10 (3 h) |
| 10 | H | Ph | IV (7.5) | 15ad | SMd | 70 (1 h) |
| 11c | Cl | Ph | IV (5) | 15bd | — | 90 (1 h) |

Encouraged by these successful results with alkyl acrylates, we next examined the less studied cross-metathesis coupling with several vinyl ketones. Starting from methyl vinyl ketone 7c, the desired enone 15ac was isolated in a good 70% yield by combining the GH-II catalyst II with microwave irradiation (Table 1, entry 7).
Phenyl vinyl ketone12 7d was next submitted to cross-metathesis with the terminal alkene 6a (Table 1, entries 8–11). The reaction was particularly reluctant under conventional heating conditions and did not work whatever the catalyst used. Contrastingly, the coupling product 15ad was easily obtained in a modest 50% yield, in only 3 hours combining microwave irradiation with GH-II catalyst II (Table 1, entry 8). Remarkably, following fully optimized conditions (i.e. 7.5 mol% of the precatalyst IV, 1 h of microwave irradiation), the expected product 15ad was produced in a 70% yield (Table 1, entry 10). This protocol was finally extended to the more reactive chloropyridine scaffold 6b, and very satisfyingly, the highly functionalized Michael acceptor 15bd was isolated with a complete E selectivity in a 90% yield (Table 1, entry 11). This result was in accordance with the demonstrated favourable effect of a chlorine electron-withdrawing C-2 substituent that reduces the Lewis basicity of the pyridinic nitrogen atom.9a
With the precursors 15 in hands, we focused on the cyclising aza-Michael step. Even though amino-enone 15ab underwent Boc-deprotection in the presence of a catalytic amount of HCl in i-PrOH at 60 °C, the RCAM did not occur. Pleasingly, microwave heating (100 °C, 200 W) efficiently reached the whole cascade in only 45 min, yielding the targeted pyrrolidines 5ab and 5ac in 50% to 65% yields (Scheme 3, eqn (1)). In addition, the best conditions for the tandem deprotection-cyclization sequence applied to 15ad were reached under ultrasound exposure for 3 hours and to 15bd by using conventional heating at 80 °C for 2 hours.
 | ||
| Scheme 3 RCAM reaction and telescoped CM-RCAM reaction. | ||
In our continuing interest for developing economically favorable synthetic procedure, we surmised that the sequential CM/RCAM process could be telescoped. Indeed, Fustero and co-workers reported diastereoselective domino cross-metathesis/aza-Michael reaction catalyzed by ruthenium complexes with Lewis acid as co-catalyst for the synthesis of piperidine, pyrrolidine and lactam derivatives.8c,e,f Directly applied to our pyridinic olefins, the cascade process using either Ti(OiPr)4 or BF3·OEt2 as Lewis acid co-catalyst failed, providing complete degradation of the starting materials. To our delight, under microwave irradiation, sequential addition of a catalytic amount of concentrated hydrochloric acid after the cross-metathesis completion (controlled by TLC), initiated the N-Boc cleavage and activated the subsequent aza-Michael induced ring closure.
Extension of these optimized conditions provided the isolation of the original pyrrolidines 5ab, 5ac, 5ad and 5bd in 50–70% yields over three steps (Scheme 3, eqn (2)). More in details, a mixture of both diastereomers was obtained. If the 2,5-cis diastereoisomer was kinetically favoured, it rapidly equilibrated in solution toward the thermodynamically more stable 2,5-trans epimer through a well-known auto-catalyzed retro-aza-Michael/aza-Michael cyclization process.4d The relative configuration of the major diastereoisomer was established by nOesy experiments and revealed a trans configuration between the both hydrogen atoms of the pyrrolidine C-2 and C-5 atoms (Scheme 3, eqn (2)).
Conclusions
We shaped a synthetic pathway allowing a short and efficient access to a series of lobeline–nicotine, pelletierine–nicotine and lobeline–nicotine–epibatidine hybrids. More generally, we demonstrated that this approach based on a challenging monotope sequential microwave-mediated cross-metathesis/cyclizing aza-Michael reaction is expandable to the diastereoselective preparation of 2,5-trans disubstituted pyrrolidines. This synthetic strategy has the advantages of (i) involving simple starting reagents and available Ru-precatalysts, (ii) producing several purification-free synthetic intermediates and (iii) facilitating the introduction of molecular diversity. Moreover, the developed methodology enabled, under microwave irradiation, the telescoping of the cross-metathesis and the intramolecular aza-Michael reaction into a single efficient process, readily amenable to scale up. New applications of this strategy for the enantioselective synthesis of pyrrolidines as well as the nAChR-subtypes binding affinity and activity of selected nicotine–lobeline hybrid analogues are currently in progress and will be reported in due course.Acknowledgements
The authors are indebted to Oméga Cat System Company for the generous gifts of ruthenium catalysts. The authors are grateful to Claire Troufflard and Karine Leblanc for performing respectively NMR experiments and elemental analyses. The authors thank the University Paris-Sud for the grant of P.-E. V. The University Paris-Sud, the LabEx LERMIT, the French Ministry of Higher Education and Research and the CNRS are gratefully acknowledged for their financial support.Notes and references
- (a) A. Taly, P. J. Corringer, D. Guedin, P. Lestage and J. P. Changeux, Nat. Rev. Drug Discovery, 2009, 8, 733 CrossRef CAS PubMed; (b) C. Gotti, F. Clementi, A. Fornari, A. A. Gaimarri, S. Guiducci, I. Manfredi, M. Moretti, P. Pedrazzi, L. Pucci and M. Zoli, Biochem. Pharmacol., 2009, 78, 703 CrossRef CAS PubMed; (c) C. Gotti, L. Riganti, S. Vailati and F. Clementi, Curr. Pharm. Des., 2006, 12, 407 CrossRef CAS PubMedA. A. Jensen, B. Frolund, T. Liljefors and P. Krogsgaard-Larsn, J. Med. Chem., 2005, 48, 4705 CrossRef CAS PubMed; (d) S. P. Arneric, M. Holladay and M. Williams, Biochem. Pharmacol., 2007, 74, 1092 CrossRef CAS PubMed; (e) M. N. Romanelli, P. Gratteri, L. Guandalini, E. Martini, C. Bonaccini and F. Gualtieri, ChemMedChem, 2007, 2, 746 CrossRef CAS PubMed.
- E. Edink, P. Rucktooa, K. Retra, A. Akdemir, T. Nahar, O. Zuiderveld, R. van Elk, E. Janssen, P. van Nierop, J. van Muijlwijk-Koezen, A. B. Smit, T. K. Sixma, R. Leurs and I. J. P. de Esch, J. Am. Chem. Soc., 2011, 133, 5363 CrossRef CAS PubMed.
- (a) A. Sutherland, T. Gallagher, C. G. V. Sharples and S. Wonnacott, J. Org. Chem., 2003, 68, 2475 CrossRef CAS PubMed; (b) E. Wright, T. Gallagher, C. G. V. Sharples and S. Wonnacott, Bioorg. Med. Chem. Lett., 1997, 7, 2867 CrossRef CAS; (c) N. Houllier, M.-C. Lasne, R. Bureau, P. Lestage and J. Rouden, Tetrahedron, 2010, 66, 9231 CrossRef CAS; (d) W. Hatton, F.-X. Felpin, M. Evain, M. Mathé-Allainmat and J. Lebreton, Synlett, 2010, 1631 Search PubMed.
- (a) L. Cabral dos Santos, Z. Bahlaouan, K. El Kassimi, C. Troufflard, F. Hendra, S. Delarue-Cochin, M. Zahouily, C. Cavé and D. Joseph, Heterocycles, 2007, 73, 751 CrossRef CAS; (b) Z. Amara, E. Drège, C. Troufflard, P. Retailleau and D. Joseph, Org. Biomol. Chem., 2012, 10, 7148 RSC; (c) H. Boufroura, M. Mauduit, E. Drège and D. Joseph, J. Org. Chem., 2013, 78, 2346 CrossRef CAS PubMed; (d) Z. Amara, G. Bernadat, P.-E. Venot, P. Retailleau, C. Troufflard, E. Drège, F. le Bideau and D. Joseph, Org. Biomol. Chem., 2014, 12, 9797 RSC; (e) E. Drège, P.-E. Venot, F. le Bideau, P. Retailleau and D. Joseph, J. Org. Chem., 2015, 80, 10119 CrossRef PubMed.
- The starting material was prepared on a 10 g scale and used without purification according to a known procedure, see: J. B. Summers, S. K. Davidsen, D. H. Steinman, J. G. Phillips, M. B. Martinand and D. E. Guinn, US Pat., 5149704, 1992.
- (a) M. A. Peterson, B. L. Nilsson, S. Sarker, B. Doboszewski, W. Zhang and M. J. Robins, J. Org. Chem., 1999, 64, 8183 CrossRef CAS PubMed; (b) Y. Pei and B. O. S. Wickham, Tetrahedron Lett., 1993, 34, 7509 CrossRef CAS.
- G. V. Reddy, G. V. Rao and D. S. Iyengar, Tetrahedron Lett., 1999, 40, 3937 CrossRef CAS.
- (a) H. Liu, C. Zeng, J. Guo, M. Zhang and S. Yu, RSC Adv., 2013, 3, 1666 RSC; (b) S.-S. P. Chou and J.-L. Huang, Tetrahedron Lett., 2012, 53, 5552 CrossRef CAS; (c) S. Fustero, C. Báez, M. Sánchez-Roselló, A. Asensio, J. Miro and C. del Pozo, Synthesis, 2012, 44, 1863 CrossRef CAS; (d) Q. Cai, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 8666 CrossRef CAS PubMed; (e) S. Fustero, S. Monteagudo, M. Sánchez-Roselló, S. Flores, P. Barrio and C. del Pozo, Chem.–Eur. J., 2010, 16, 9835 CrossRef CAS PubMed; (f) S. Fustero, D. Jiménez, M. Sánchez-Roselló and C. del Pozo, J. Am. Chem. Soc., 2007, 129, 6700 CrossRef CAS PubMed.
- (a) K. Lafaye, L. Nicolas, A. Guérinot, S. Reymond and J. Cossy, Org. Lett., 2014, 16, 4972 CrossRef CAS PubMed; (b) S. J. P'Pool and H.-J. Schanz, J. Am. Chem. Soc., 2007, 129, 14200 CrossRef PubMed.
- (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed; (b) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS; (c) H. Clavier, C. A. Urbina-Blanco and S. P. Nolan, Organometallics, 2009, 28, 2848 CrossRef CAS.
- (a) H. Clavier, F. Caijo, E. Borre, D. Rix, F. Boeda, S. P. Nolan and M. Mauduit, Eur. J. Org. Chem., 2009, 4254 CrossRef CAS; (b) D. Rix, F. Caijo, I. Laurent, F. Boeda, H. Clavier, S. P. Nolan and M. Mauduit, J. Org. Chem., 2008, 73, 4225 CrossRef CAS PubMed.
- For the preparation of 7d, see: F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20930g |
| This journal is © The Royal Society of Chemistry 2015 |