
Triisopropylsilylacetylene-functionalised anthracene-alt-benzothiadiazole copolymers for application in bulk heterojunction solar cells†
Abstract
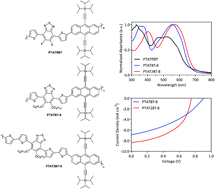
Three triisopropylsilylacetylene-functionalised anthracene (TIPSAnt) based polymers were synthesised by copolymerising TIPSAnt with either dithienyl-5,6-difluoro-benzo[c]-[1,2,5]thiadiazole, dithienyl-benzo[c]-[1,2,5]thiadiazole or dibithiophenyl-benzo[c]-[1,2,5]thiadiazole to yield PTATffBT, PTATBT-8 and PTAT2BT-8, respectively. PTAT2BT-8 demonstrated a reduced optical and electrochemical band gap, relative to PTATffBT and PTATBT-8. The HOMO level of PTAT2BT-8 (−5.32 eV) is significantly shallower compared to its counterparts. This can be attributed to increased intramolecular charge transfer along the polymer backbone; a consequence of the incorporation of additional thiophene spacer units. The photovoltaic properties of the polymers were investigated by fabricating bulk heterojunction (BHJ) polymer solar cells using PC70BM as the electron acceptor. PTATffBT displayed limited solubility in common organic solvents and could not be used for the fabrication of photovoltaic cells. Optimised photovoltaic devices fabricated from PTATBT-8 and PTAT2BT-8 demonstrated power conversion efficiencies of 2.36% and 3.15%, respectively. PTAT2BT-8 provided better efficiencies chiefly as a result of better Jsc and FF values.
 Please wait while we load your content...
Please wait while we load your content...

