
Scaling of optical forces on Au–PEG core–shell nanoparticles
Abstract
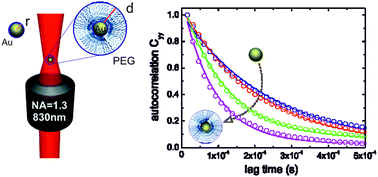
Optical trapping of hybrid core–shell gold–polymer particles is studied. Optical forces are measured for different gold core size and polymer shell thickness, revealing how a polymer shell increases the trapping efficiency with respect to the bare gold nanoparticles. Data are in agreement with calculations of optical trapping based on electromagnetic scattering theory in the T-matrix approach. The scaling behaviour of optical forces with respect to the ratio between polymer layer thickness and the whole particle radius is found and discussed.
 Please wait while we load your content...
Please wait while we load your content...

