
Crosslinked polyurea aerogels with controlled porosity
Abstract
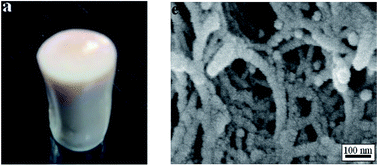
Mechanically robust polyurea aerogels with controlled porosity are synthesized from aromatic isocyanates, aromatic diamines, and a triamine crosslinker. Linear, isocyanate end-capped polyurea oligomers are first synthesized from the reactions between the diamine and 4,4′-diphenylmethane diisocyanate in anhydrous N-methyl-2-pyrrolidone (NMP). The oligomers are then cross-linked with 1,3,5-triaminophenoxylbenzene to produce the gels. The gels are dried under supercritical conditions of carbon dioxide after exchanging NMP with acetone and acetone with liquid carbon dioxide. The aerogels are mesoporous with mean pore diameter in the range of 9–16 nm, have a bulk density of 0.19–0.26 g cm−3, porosity of 79–86%, and surface areas between 106 and 309 m2 g−1. Pore size distributions broaden and shift to larger diameter as the crosslink density is reduced. The spectroscopic evidence suggests that hydrogen bonding is effective in reducing the shrinkage of aerogels, especially when linear oligomers of higher molecular weight are used. These materials show onset of thermal decomposition at 250 °C and offer high compressive moduli in the range of 12–69 MPa.
 Please wait while we load your content...
Please wait while we load your content...

