
Simple and convenient G-quadruplex-based fluorescence assay of micrococcal nuclease activity†
Yue He*ab and
Bining Jiao*ab
aLaboratory of Quality & Safety Risk Assessment for Citrus Products (Chongqing), Ministry of Agriculture, Citrus Research Institute, Southwest University, Chongqing, 400712, China
bNational Citrus Engineering Research Center, Chongqing, 400712, China. E-mail: yhe@swu.edu.cn; Fax: +86 23 68349603; Tel: +86 23 68349603
First published on 3rd December 2015
Abstract
As the extracellular nuclease of Staphylococcus aureus (S. aureus), micrococcal nuclease (MNase), which preferentially digests single-stranded nucleic acids, can be used as the standard to identify S. aureus and can be used to evaluate the pathogenicity of S. aureus. So the assay of MNase is of high importance. However, traditional methods for the assay of MNase activity have intrinsic limitations such as sophisticated synthesis processes, the need for functionalizing thiol (or dye)-modified oligonucleotide probes or critical operation conditions. Herein, a simple and convenient fluorescence sensing platform for MNase activity has been developed based on N-methyl mesoporphyrin IX (NMM)/G-quadruplexes. In the absence of MNase, the G-rich single stranded DNA (ssDNA) folds into a quadruplex in the presence of monovalent ions, thus greatly enhancing the fluorescence of NMM (a specific G-quadruplex binder). In the presence of MNase, the G-rich ssDNA was digested into small fragments. As a result, the fluorescence of the solution decreases with increase of MNase activity. Under the optimized conditions, the fluorescence intensity exhibits a linear correlation to MNase concentration in a wide range of 1.2 × 10−4–2.4 × 10−3 units per mL with a detection limit of 7.1 × 10−5 units per mL and good selectivity. The detection limit is much better or at least comparable to previous reports. Given its simplicity, easy operation, sensitivity and cost-effectiveness, this method can be extended to other nuclease assays.
Introduction
Micrococcal nuclease (MNase), as a nonspecific endo–exonuclease, digests single- and double-stranded DNA and RNA. But it preferentially digests single-stranded DNA (ssDNA).1,2 MNase is a thermostable nuclease, its activity is strictly dependent on Ca2+.3 MNase has been used in hydrolysis of nucleic acids, sequencing of RNA, and studies of chromatin and protein structure.4–7 In addition, the existence of MNase can be a standard to identify S. aureus and the content of MNase can be used to evaluate the pathogenicity of S. aureus.8,9 Traditional techniques, including gel electrophoresis,10,11 high-performance liquid chromatography (HPLC),12 enzyme-linked immunosorbent assay (ELISA)13 and radioactive labeling,14 have been established to monitor nuclease activity. However, most of them are time-consuming, laborious, and require sophisticated instrumentation. To address these limitations, fluorescence assays have been applied and developed because of their intrinsic high sensitivity and selectivity. Among these fluorescence method, quantum dots (QDs) and silver nanoclusters (AgNCs) have often been used in MNase detection assays. He et al. firstly developed a fluorescence resonance energy transfer (FRET) method for the detection of MNase activity.15 In this method, they modified the ssDNA substrates with QDs at the 3′-end and 6-carboxy-X-rhodamine (ROX) at the 5′-end, respectively. FRET between QDs and ROX resulted in a color change of the system from green to orange-red. After the ssDNA was cleaved by MNase into small fragments, the color changed back to green. However, this strategy is compromised to expensive double fluorophore-labeled DNA substrates. Subsequently, in order to avoid this shortcoming, they reported another QDs-based FRET probe for MNase detection by means of the electrostatic principle.16 However, this method involved a modification process to get positively charged QDs via two-step conjugation reaction, and the process was complicated and time-intensive. To further reduce the modification, Jiang et al. developed a QDs-FRET system through peptide acting as an electrostatic linker.17 Although promising, these strategies involved the use of expensive fluorophore-labeled DNA substrates and they can suffer from problems associated with high background signal, due to the low energy transfer efficiency between the donor and the acceptor. Recently, Jiang et al. developed a label-free method for MNase detection using AgNCs as the signal reporter.18 This strategy did not require any fluorescence dye label or a complex design. However, the need for AgNCs synthesis procedures added to the complexity. Therefore, it is still highly desirable to develop facile label-free methods to monitor MNase activity.Recently, G-quadruplexes which formed by G-rich DNA or RNA sequences have been received intense research interest as optical sensing elements.19–21 Due to their structure polymorphism and tunable conformation, G-quadruplexes have been widely used for constructing biosensors for the colorimetric,22,23 chemiluminescence,24,25 or fluorescent26–28 detection of a series of analytes. For instance, Wang's group developed a G-quadruplex-based fluorescent method for S1 nuclease activity and K+ assays with protoporphyrin IX (PPIX) as a signal reporter.28 In their work, they find out that G-quadruplex DNA can greatly enhance the fluorescence of PPIX. With the digestion of G-quadruplex DNA by S1 nuclease, emission response of PPIX decreased. Furthermore, they find out that K+ can reduce the activity of S1 nuclease towards G-quadruplex DNA. Based on these findings, this G-quadruplex-based biosensor showed high sensitivity for S1 nuclease and K+. In addition to metal ion K+, proteins and small molecules also can reduce the activity of nuclease toward DNA.29 Based on these findings, Dong's group developed another G-quadruplex-based fluorescent method for ATP assays with berberine as the signal reporter.30 These facile label-free G-quadruplex-based biosensors are highly selective and sensitive. Among all the G-quadruplex signal reporters, N-methyl mesoporphyrin IX (NMM) has been frequently used to construct biosensors for various target molecules detection.31–35 NMM is a commercially available unsymmetrical anionic porphyrin characterized by a pronounced structural selectivity for G-quadruplex but not for duplexes, triplexes, or single-stranded forms.36,37 The fluorescence of NMM is very weak, but exhibits a 20-fold enhancement upon interacting with the G-quadruplex DNA structure (with excitation and emission wavelengths centered at 399 and 608 nm, respectively).31–35 NMM was reported to interact with a variety of G-quadruplex forms, among them d[G3(T4G3)3] gave the best fluorescence enhancement.34
In the present work, MNase is found to destroy monovalent cation-stabilized G-quadruplexes, and the fluorescent response of NMM is dependent on the concentration of MNase. Inspired by these phenomena, we expect that the combination of signal responsive G-quadruplexes/NMM complexes and cleavage reactions could offer a unique opportunity to construct robust sensor for MNase. Significantly, such system does not require sophisticated design or chemical labeling, which reduces complexity, cost and overall assay time. Meanwhile, this strategy might pave the way to apply G-quadruplexes as novel transducers for more nuclease sensing systems.
Experimental section
Chemicals and materials
The ssDNA d[G3(T4G3)3] with a sequence of 5′-GGGTTTTGGGTTTTGGGTTTTGGG-3′ was synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China).N-Methylmesoporphyrin IX (NMM) was purchased from J&K Scientific Ltd. (Beijing, China). Micrococcal nuclease (MNase), exonuclease I (Exo I), exonuclease III (Exo III), and deoxyribonuclease I (DNase I) were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). S1 nuclease was purchased from Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). The Tris–HCl buffer used in this experiment consisted of 20 mM Tris–HCl (pH 8.0), 5 mM NaCl and 2.5 mM CaCl2. Milli-Q purified water was used to prepare all the solutions.
Apparatus
Fluorescent emission spectra were performed on Varian cary eclipse fluorescence spectrophotometer, Varian Medical Systems, Inc. (Palo Alto, American). The sample cell is a 700 μL quartz cuvette. The luminescence intensity was monitored by exciting the sample at 399 nm and measuring the emission at 608 nm. The slits for excitation and emission were set at 5 nm, 10 nm respectively. The fitting of the experimental data was accomplished using the software Origin 8.0.Optimization of the concentration of d[G3(T4G3)3]
Tris–HCl buffer (MNase working buffer) was used to prepare all the solutions. To optimize the concentration of ssDNA d[G3(T4G3)3], 0, 2, 4, 6, 8, and 10 μL d[G3(T4G3)3] solution (100 μM) and 20 μL of the NMM stock solution (20 μM) were mixed. Then appropriate concentration of KCl solution were added and the final volume of the solution was 500 μL. Finally, the above prepared solution was incubated 10 min at room temperature. The fluorescence intensity of the incubated solution was measured at 608 nm with excitation at 399 nm.Optimization of the concentration of KCl
Tris–HCl buffer (MNase working buffer) was used to prepare all the solutions. To optimize the concentration of KCl, 10 μL of the ssDNA d[G3(T4G3)3] stock solution (100 μM) and 20 μL of the NMM stock solution (20 μM) were mixed. Then 0, 2.5, 5, 12.5, 25, 50, 75 and 100 μL KCl solution (100 mM) as prepared were added and the final volume of the solution was 500 μL. Finally, the above prepared solution was incubated 10 min at room temperature. The fluorescence intensity of the incubated solution was measured at 608 nm with excitation at 399 nm.Optimization of the reaction time between ssDNA d[G3(T4G3)3] and MNase
Tris–HCl buffer (MNase working buffer) was used to prepare all the solutions. To optimization the reaction time between ssDNA d[G3(T4G3)3] and MNase assays, 10 μL of the ssDNA d[G3(T4G3)3] stock solution (100 μM) and 1.2 × 10−3 units per mL MNase solution were mixed. The above prepared solution was incubated for 0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, and 60 min at 37 °C. Then 20 μL of the NMM stock solution (20 μM) and 25 μl KCl solution (100 mM) were added. The mixed solution was diluted with Tris–HCl buffer to 500 μL. Finally, the above prepared solution was incubated 10 min at room temperature. The fluorescence intensity of the incubated solution was measured at 608 nm with excitation at 399 nm.Performance of MNase detection
Tris–HCl buffer (MNase working buffer) was used to prepare all the solutions. For MNase assays, 10 μL of the ssDNA d[G3(T4G3)3] stock solution (100 μM) and appropriate concentrations of MNase solution were mixed. The above prepared solution was incubated for 30 min at 37 °C. Then 20 μL of the NMM stock solution (20 μM) and 25 μl KCl solution (100 mM) were added. The mixed solution was diluted with Tris–HCl buffer to 500 μL. Finally, the above prepared solution was incubated 10 min at room temperature. The fluorescence intensity of the incubated solution was measured at 608 nm with excitation at 399 nm.Performance of MNase detection in culture medium
Culture medium (10-fold diluted with Tris–HCl buffer) was used to prepare all the solutions. For MNase in culture medium assays, 10 μL of the ssDNA d[G3(T4G3)3] stock solution (100 μM) and appropriate concentrations of MNase solution were mixed. The above prepared solution was incubated for 30 min at 37 °C. Then 20 μL of the NMM stock solution (20 μM) and 25 μl KCl solution (100 mM) were added. The mixed solution was diluted with culture medium solution to 500 μL. Finally, the above prepared solution was incubated 10 min at room temperature. The fluorescence intensity of the incubated solution was measured at 608 nm with excitation at 399 nm.Results and discussion
Design strategy
Fig. 1 illustrates the sensing strategy for the detection of MNase. In the absence of MNase, the ssDNA d[G3(T4G3)3] was not digested. It forms of the G-quadruplex structure after the addition of KCl. Therefore, through the strong interaction between the G-quadruplex structure and NMM, a significant fluorescence enhancement of NMM can be detected. However, upon the addition of MNase, the d[G3(T4G3)3] is cut into fragments by MNase, hampering the formation of the G-quadruplex structure in the presence of KCl. Therefore, no fluorescence enhancement can be detected. | ||
| Fig. 1 Scheme for the mechanism of G-quadruplex-based biosensor for MNase detection. | ||
To demonstrate the feasibility of this approach, the process of fluorescence changing of the G-quadruplex-based biosensor for MNase detection is shown by fluorescence spectra. Fig. 2 shows the fluorescence emission spectra of NMM under different conditions. The fluorescence spectrum of NMM alone (Fig. 2, curve a) in Tris–HCl buffer shows week fluorescence intensity. However, a significant increase (17-fold) in fluorescence intensity was observed upon addition of d[G3(T4G3)3] in the presence of KCl (Fig. 2, curve b) because of the strong interaction between NMM and the G-quadruplex structure. Upon the addition of MNase (1.2 × 10−3 units per mL) to d[G3(T4G3)3] solution, and incubating for 30 min at 37 °C. The fluorescence intensity of NMM decreased significantly (Fig. 2, curve c), which means cleavage reaction appeared between MNase and d[G3(T4G3)3]. Therefore, MNase detection could be easily realized by monitoring the change of fluorescence signal.
 | ||
| Fig. 2 Fluorescence emission spectra of NMM under different conditions: (a) NMM; (b) d[G3(T4G3)3] + NMM; (c) d[G3(T4G3)3] + MNase + NMM. Concentration: NMM, 0.8 μM; d[G3(T4G3)3], 2 μM; MNase, 1.2 × 10−3 units per mL. Excitation: 399 nm. | ||
Effect of K+
In order to achieve the sensing performance, we first investigate the influence of the concentrations of K+. It is well-known that K+ can be used as a stabilizer for G-quadruplexes formation. As we can see from Fig. 3, the fluorescence intensity of NMM in d[G3(T4G3)3] solution increased gradually as the concentration of KCl increased. This result is in accordance with Hu's work.34 And we take 5 mM as the optimized concentration for K+. Other reports reported that metal ion such as K+, proteins and small molecules can reduce the activity of nuclease toward DNA.28,29 So, next, we investigate the influence of the adding order of MNase and K+ on the final results (ESI, Fig. S1†). ESI, Fig. S1,† curve b shows that considerably low fluorescence signal of the system appears when we first make the digestion of d[G3(T4G3)3] by MNase happening before the addition of K+. In contrast, in Fig. S1,† curve c, which shows the result of the reversed adding order of MNase and K+ (ESI, Fig. S1,† curve b), still exhibits very high fluorescence signal, which means little d[G3(T4G3)3] has been digested by MNase in this condition. Therefore the adding order of MNase and K+ has significant influence on the final results. This phenomenon can be explained by the properties of K+ which can protect G-quadruplex against enzymatic cleavage.28 Besides, the protection effect increases with the increasing concentration of K+ (ESI, Fig. S2†). As a result, to achieve the sensing performance of MNase detection, MNase should be added before the addition of K+.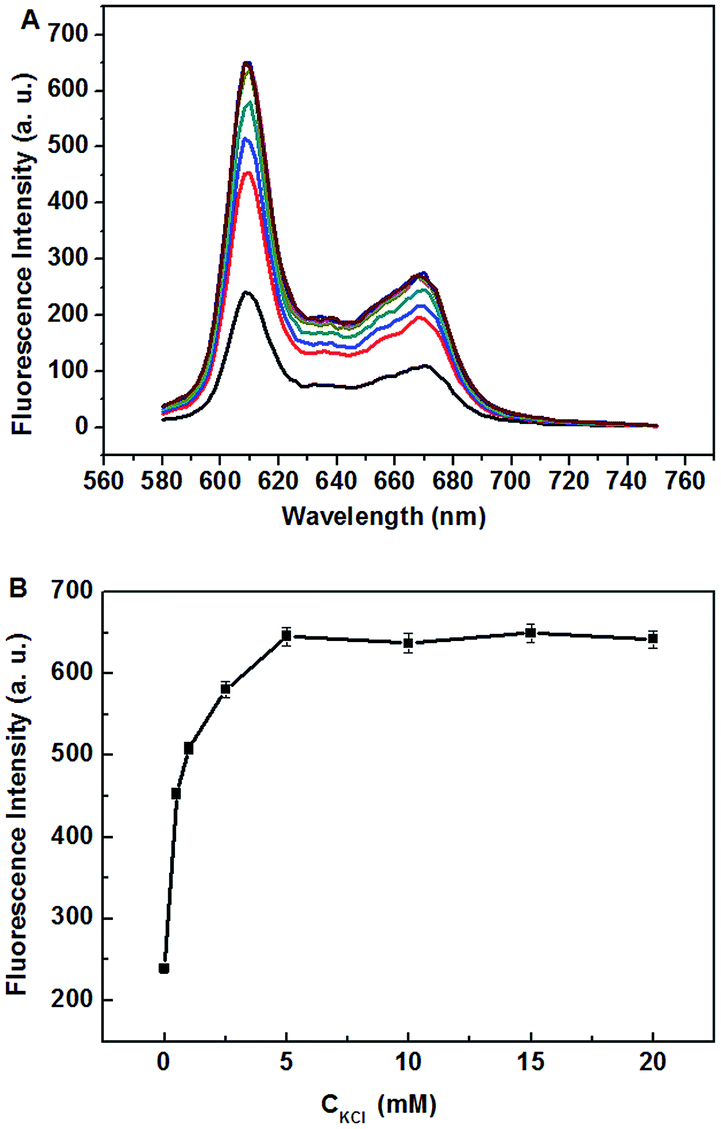 | ||
| Fig. 3 Fluorescence emission spectra of NMM in the presence of d[G3(T4G3)3] and increasing amount of K+ (A) and fluorescence intensity of NMM in the presence of d[G3(T4G3)3] and different concentration of K+ (B). Excitation: 399 nm. | ||
Optimization of detection strategy
In order to establish optimal conditions for the fluorescence assay, relevant experimental parameters affecting the reaction, including the option of the concentration of d[G3(T4G3)3], and MNase-catalyzed digestion reaction time were assessed and optimized.To achieve the best sensing performance, the concentration of d[G3(T4G3)3] was optimized. As shown in Fig. 4, fluorescence intensity of NMM increases sharply as the concentration of d[G3(T4G3)3] increases. Moreover, there is a good linear relationship between the fluorescence intensity of NMM and the concentration of d[G3(T4G3)3] from 0 to 2 μM. As a result, 2 μM was taken as the optimized concentration for d[G3(T4G3)3].
 | ||
| Fig. 4 Fluorescence emission spectra of NMM in the presence of increasing amount of d[G3(T4G3)3] (A) and linear relationship between fluorescence intensity of NMM and the concentration of d[G3(T4G3)3] (B). Excitation: 399 nm. | ||
The kinetic behaviors of d[G3(T4G3)3] and MNase was studied by monitoring the fluorescence intensity as a function of time. Fig. 5 shows fluorescence intensity versus different reaction time of d[G3(T4G3)3] (2 μM) with MNase (1.2 × 10−3 units per mL). It indicates that the reaction of d[G3(T4G3)3] with MNase is very fast and is completed in about 30 min at 37 °C. So we take 30 min as the optimized reaction time.
 | ||
| Fig. 5 Fluorescence decreasing of the G-quadruplex-based biosensor by MNase as a function of time. Concentration: NMM, 0.8 μM; d[G3(T4G3)3], 2 μM; MNase, 1.2 × 10−3 units per mL. Excitation: 399 nm. | ||
MNase detection
The assay of MNase was carried out under the optimized conditions with the fixed concentrations of NMM (0.8 μM) ssDNA d[G3(T4G3)3] (2 μM) and KCl (5 mM). Fig. 6A shows the fluorescence emission spectra of the G-quadruplex-based biosensor in the presence of different concentrations of MNase. The fluorescence intensity of the biosensor dramatically decreases with the increasing concentration of MNase. The calibration curve for MNase detection is shown in Fig. 6B, and the linear range is from 1.2 × 10−4–2.4 × 10−3 units per mL with linear equation y = −238119.63x + 620.07, where y is the fluorescence intensity of NMM at 608 nm and x is the concentration of MNase (determination coefficient R2 = 0.9888). The detection limit is estimated to be 7.1 × 10−5 units per mL (3S0/S, in which S0 is the standard deviation for the blank solution, n = 11, and S is the slope of the calibration curve), which is much better or at least comparable to previous reported MNase biosensors.15–18,38 A series of eleven repetitive measurements of 1.2 × 10−3 units per mL MNase were used for estimating the precision, and the relative standard deviation (RSD) was 3.4%, showing good reproducibility of the proposed method. | ||
| Fig. 6 Fluorescence emission spectra of G-quadruplex-based biosensor in the presence of increasing amount of MNase (A) and fluorescence intensity of G-quadruplex-based biosensor in the presence of different concentration of MNase (inset: calibration curve for MNase detection) (B). Excitation: 399 nm. | ||
Specificity
In this study, under the optimized detection condition, other nucleases (Exo I, S1 nuclease, DNase I and Exo III) were selected to study the specificity of G-quadruplex-based biosensor under the same conditions. Exo I breaks apart single-stranded DNA in the direction 3′ → 5′, releasing deoxyribonucleoside 5′-monophosphates one after another. Exo I has its maximal activity at the basic pH (9.5) and requires Mg2+ for maximal activity. The S1 nuclease also degrades single-stranded nucleic acids, releasing 5′-phosphoryl mono- or oligonucleotides. The excision of ssDNA by S1 nuclease has also occurred in AT-rich regions, which is similar to the cleavage site of MNase.39 But, it has been reported that the S1 nuclease has its maximal activity at the acid pH (4.2) and requires Zn2+ or Co2+ for maximal activity.40 Exo III is a 3′ → 5′ exonuclease specific for dsDNA or DNA–RNA hybrids. In a typical experiment, ssDNA d[G3(T4G3)3] was mixed with 1.2 × 10−3 units per mL MNase, Exo I, S1 nuclease, DNase I and Exo III for 30 min at 37 °C in Tris–HCl buffer (pH 8.0), respectively. And then NMM and KCl solution were added and incubated 10 min at room temperature. As shown in Fig. 7, the fluorescence intensity of this G-quadruplex-based biosensor changed less for Exo I, S1 nuclease, DNase I and Exo III, while a significant fluorescence decrease was observed for MNase. The observation indicates the specificity of the G-quadruplex-based biosensor for MNase testing. | ||
| Fig. 7 The relative fluorescence F0 − F of G-quadruplex-based biosensor in the presence of different nucleases: blank control (without MNase); MNase (1.2 × 10−3 units per mL); Exo I (1.2 × 10−3 units per mL); S1 nuclease (1.2 × 10−3 units per mL), DNase I (1.2 × 10−3 units per mL), and Exo III (1.2 × 10−3 units per mL). Excitation: 399 nm. | ||
MNase detection in culture medium
In order to evaluate the practicability of the proposed design, the detection of MNase activity in bacteria culture medium, a kind of fluid sample with complicated biological matrix, was also performed. The detection process of MNase in 10% culture medium was the same as in the Tris–HCl buffer. As shown in ESI, Fig. S2A,† the fluorescence intensity of the biosensor still dramatically decreases with the increasing concentration of MNase. And there is still a good linear relationship between the fluorescence intensity and the concentration of MNase from 1.2 × 10−4 to 2.4 × 10−3 units per mL (ESI, Fig. S2B†). These results clearly demonstrate this G-quadruplex-based biosensor can be used to detect MNase in practical samples sensitively.Conclusions
In summary, a simple and ultrasensitive strategy for micrococcal nuclease (MNase) detection based on G-quadruplex is developed in this work. The commercial chemical reagent N-methyl mesoporphyrin IX (NMM) was chosed as the signal element as its good selectivity for G-quadruplex but not for duplexes, triplexes, or single-stranded forms. The fluorescence of NMM is very weak, but exhibits a 17-fold enhancement upon interacting with the G-quadruplex DNA structure. Upon the addition of MNase into the G-quadruplex DNA solution, the G-quadruplex DNA was cleaved into small fragments. The fluorescence intensity dramatically decreased with the increasing concentration of MNase. Therefore, the emission intensity changes could be directly related to the amount of MNase added to the assay solution. This G-quadruplex-based biosensor is extraordinarily sensitive to MNase detection. As to MNase, a sensitive detection limit of 7.1 × 10−5 units per mL was obtained. In addition, the new strategy is featured with high simplicity. Therefore, we can reasonably expect that the G-quadruplex-based biosensor model can be readily developed for other nucleases whose substrates are ssDNA molecules such as S1 nuclease just by changing the reaction buffer.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21405125), Natural Science Foundation Project of CQ (No. CSTC2014JCYJA80041), Fundamental Research Funds for the Central Universities (No. XDJK2015C090 and SWU113099), the National Key Technology R&D Program (No. 2009BADB7B04) and China Agriculture Research System (No. CARS-27).Notes and references
- K. R. Fox and M. J. Waring, Biochim. Biophys. Acta, 1987, 909, 145–155 CrossRef CAS.
- T. M. Martensen and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 6458–6460 CrossRef CAS.
- P. Cuatreca, S. Fuchs and C. B. Anfinsen, J. Biol. Chem., 1967, 242, 1541–1547 Search PubMed.
- S. B. Svetlikova and V. A. Pospelov, Mol. Biol. Rep., 1986, 20, 160–166 Search PubMed.
- S. P. Modak and P. Beard, Nucleic Acids Res., 1980, 8, 2665–2678 CrossRef CAS PubMed.
- M. Wal and B. F. Pugh, in Nucleosomes, Histones & Chromatin, Pt B, eds. C. Wu and C. D. Allis, Elsevier Academic Press Inc, San Diego, 2012, vol. 513, pp. 233–250 Search PubMed.
- T. R. Butt, D. B. Jump and M. E. Smulson, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1628–1632 CrossRef CAS.
- A. Arnone, C. J. Bier, F. A. Cotton, E. E. Hazen, D. C. Richards and J. S. Richards, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 420–427 CrossRef CAS.
- P. R. S. Lagae-Wiens, M. J. Alfa, K. Manickam and J. A. Karlowsky, J. Clin. Microbiol., 2007, 45, 3478–3479 CrossRef PubMed.
- A. L. Rosenthal and S. A. Lacks, Anal. Biochem., 1977, 80, 76–90 CrossRef CAS PubMed.
- T. Karpetsky, G. E. Brown, E. Mcfarland, S. T. Brady, W. Roth, A. Rahman and P. Jewett, Biochem. J., 1984, 219, 553–561 CrossRef CAS PubMed.
- K. Hiramatsu, H. Miura, S. Kamei, K. Iwasaki and M. Kawakita, J. Biochem., 1998, 124, 231–236 CrossRef CAS PubMed.
- A. Jeltsch, A. Fritz, J. Alves, H. Wolfes and A. Pingoud, Anal. Biochem., 1993, 213, 234–240 CrossRef CAS.
- G. S. Boswell, S. D. Dimitrijevich and R. W. Gracy, Anal. Biochem., 1989, 182, 20–24 CrossRef CAS.
- S. Huang, Q. Xiao, Z. K. He, Y. Liu, P. Tinnefeld, X. R. Su and X. N. Peng, Chem. Commun., 2008, 45, 5990–5992 RSC.
- T. Qiu, D. Zhao, G. H. Zhou, Y. A. Liang, Z. K. He, Z. H. Liu, X. N. Peng and L. Zhou, Analyst, 2010, 135, 2394–2399 RSC.
- Y. H. Chen, L. Wang and W. Jiang, Talanta, 2012, 97, 533–538 CrossRef CAS PubMed.
- Y. L. Peng, J. H. Jiang and R. Q. Yu, Anal. Methods, 2014, 6, 4090–4094 RSC.
- C. C. Hardin, T. Watson, M. Corregan and C. Bailey, Biochemistry, 1992, 31, 833–841 CrossRef CAS PubMed.
- W. Guschlbauer, J. F. Chantot and D. Thiele, J. Biomol. Struct. Dyn., 1990, 8, 491–511 CAS.
- T. Simonsson, Biol. Chem., 2001, 382, 621–628 CrossRef CAS PubMed.
- T. Li, S. J. Dong and E. Wang, Anal. Chem., 2009, 81, 2144–2149 CrossRef CAS PubMed.
- T. Li, E. Wang and S. J. Dong, Chem. Commun., 2009, 580–582, 10.1039/b815814b.
- R. Freeman, X. Q. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133, 11597–11604 CrossRef CAS PubMed.
- X. Q. Liu, R. Freeman, E. Golub and I. Willner, ACS Nano, 2011, 5, 7648–7655 CrossRef CAS PubMed.
- Z. X. Zhang, E. Sharon, R. Freeman, X. Q. Liu and I. Willner, Anal. Chem., 2012, 84, 4789–4797 CrossRef CAS PubMed.
- H. Ueyama, M. Takagi and S. Takenaka, J. Am. Chem. Soc., 2002, 124, 14286–14287 CrossRef CAS.
- Z. X. Zhou, J. B. Zhu, L. B. Zhang, Y. Du, S. J. Dong and E. K. Wang, Anal. Chem., 2013, 85, 2431–2435 CrossRef CAS PubMed.
- D. M. Zheng, R. X. Zou and X. H. Lou, Anal. Chem., 2012, 84, 3554–3560 CrossRef CAS PubMed.
- Y. L. Wei, Y. X. Chen, H. H. Li, S. M. Shuang, C. Dong and G. F. Wang, Biosens. Bioelectron., 2015, 63, 311–316 CrossRef CAS PubMed.
- J. J. Zhao, C. F. Chen, L. L. Zhang, J. H. Jiang, G. L. Shen and R. Q. Yu, Analyst, 2013, 138, 1713–1718 RSC.
- J. T. Ren, H. X. Qin, J. H. Wang, N. W. Luedtke, E. K. Wang and J. Wang, Anal. Bioanal. Chem., 2011, 399, 2763–2770 CrossRef CAS PubMed.
- D. Hu, Z. Z. Huang, F. Pu, J. S. Ren and X. G. Qu, Chem. –Eur. J., 2011, 17, 1635–1641 CrossRef CAS.
- D. Hu, F. Pu, Z. Z. Huang, J. S. Ren and X. G. Qu, Chem. –Eur. J., 2010, 16, 2605–2610 CrossRef CAS PubMed.
- C. Q. Zhao, L. Wu, J. S. Ren and X. G. Qu, Chem. Commun., 2011, 47, 5461–5463 RSC.
- J. S. Ren and J. B. Chaires, Biochemistry, 1999, 38, 16067–16075 CrossRef CAS PubMed.
- H. Arthanari, S. Basu, T. L. Kawano and P. H. Bolton, Nucleic Acids Res., 1998, 26, 3724–3728 CrossRef CAS.
- Y. He, L. H. Xiong, X. J. Xing, H. W. Tang and D. W. Pang, Biosens. Bioelectron., 2013, 42, 467–473 CrossRef CAS PubMed.
- H. Hofstetter, A. Schambock, J. Vandenberg and C. Weissmann, Biochim. Biophys. Acta, 1976, 454, 587–591 CrossRef CAS.
- V. M. Vogt, Eur. J. Biochem., 1973, 33, 192–200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20773h |
| This journal is © The Royal Society of Chemistry 2015 |