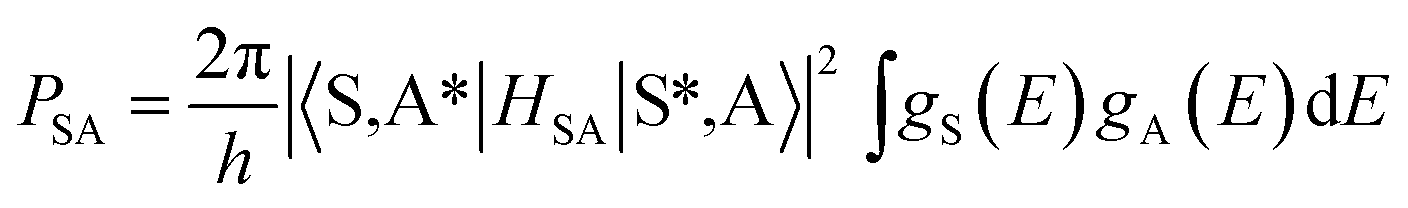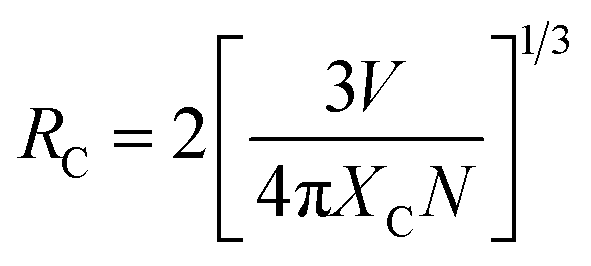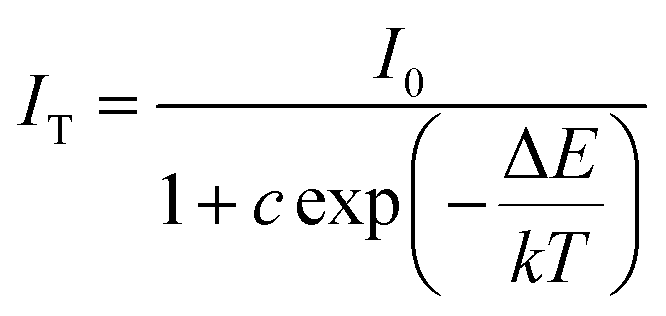DOI:
10.1039/C5RA20756H
(Paper)
RSC Adv., 2015,
5, 98350-98360
Structure, luminescence properties and energy transfer behavior of color-adjustable Sr3Gd2(Si3O9)2:Ce3+, Tb3+/Mn2+ phosphors†
Received
7th October 2015
, Accepted 9th November 2015
First published on 11th November 2015
Abstract
A series of single-phased and emission-tunable Sr3Gd2(Si3O9)2 (SGSO):Ce3+, Tb3+/Mn2+ phosphors have been successfully prepared via the solid-state reaction, and the crystal structures, luminescence properties, energy transfer of Ce3+ → Tb3+ and Ce3+ → Mn2+, color tuning and thermally stability were systematically investigated respectively. The energy transfer from Ce3+ to Tb3+ or Mn2+ ions were deduced from the spectral overlap between the Ce3+ emission and Tb3+/Mn2+ excitation spectra. The energy transfer mechanisms of Ce3+ → Tb3+ and Ce3+ → Mn2+ in the host were verified to be a dipole–quadrupole interaction and dipole–dipole interaction, respectively, which made the emission color shift from blue to green and near white with the corresponding Commission Internationale deL'Eclairage (CIE) chromaticity coordinates from (0.174, 0.060) to (0.286, 0.615) and (0.319, 0.367), respectively. The good thermal stability of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.45Mn2+ samples showed about 72.3% and 86.1% at 150 °C of its initial emission intensity at room temperature due to the different energy transfer efficiency through Ce3+ → Tb3+ and Ce3+ → Mn2+. The maximum quantum yields (QYs) of as-prepared SGSO:0.26Ce3+, 0.10Tb3+ and SGSO:0.26Ce3+, 0.57Mn2+ phosphors could reach 80.2% and 62.4%, respectively. All these properties indicate that the SGSO:Ce3+, Tb3+/Mn2+ phosphors have potential applications as ultraviolet-convertible phosphors.
1. Introduction
In recent years, most of the interest in luminescent rare-earth ions has concentrated on the single-phase multi-color emission phosphors.1–3 Due to their unique properties including but not limited to stability of color temperature, good color rendering index and low cost, single-phased multi-color emission phosphors are key to designing novel high-performance phosphors for the indispensable solid-state light source of phosphor-converted devices, such as fluorescence lamps, white light emitting diodes (WLEDs), and long-lasting phosphorescence materials, and so on.4–6
To generate multi-color emission from single-composition phosphors, two strategies have generally been adopted to modify the phosphors. One of the strategies is a modification of the composition of one activator ion doped the host by creating additional “sites” for this ion with another shift emission while retaining the first emission in the same host.7,8 For example, the emission of Ce3+ in Lu2CaMg2Si3O12 can be strongly red-shifted when there is a larger crystal field splitting of the two lowest-energy 5d levels, or creating additional sites for Ce3+ with red emission via incorporating Si4+–N3+ into the YAG:Ce3+ lattice.9,10 Another approach leading to multi-color emission in a single host is by co-doping sensitizer and activator into a crystalline matrix, using the principle of energy transfer from sensitizer to activator,11 such as Eu2+/Mn2+, Eu2+/Tb3+, Ce3+/Eu2+ Ce3+/Mn2+ and Ce3+/Tb3+, which have been investigated in many hosts. For instance, La6Ba4(SiO4)6F2:Ce3+, Tb3+,12 Na2CaMg(PO4)2:Eu2+, Mn2+,13 Ca3Si2O7:Ce3+, Eu2+,14 Na2Ca4(PO4)3F:Eu2+, Tb3+,15 Sr2MgSi2O7:Eu2+, Tb3+,16 NaBa4(BO3)3:Ce3+, Tb3+,17 Y4Si2O7N2:Ce3+, Tb3+,18 Sr7La3[(PO4)2.5(SiO4)3(BO4)0.5](BO2):Ce3+, Mn2+,19 Na4Ca4Si6O18:Ce3+, Mn2+.20
Incorporating two different luminescent centers into the host is the most effective way to broaden the applicability of inorganic phosphors. Because the Ce3+ ion commonly shows efficient broad band emission due to the 4f–5d parity allowed electric dipole transition, and it has a larger Stokes shift than those of the other RE ions owing to the extended radial wave functions of the 5d state.21 The Ce3+ ion can act as an efficient sensitizer by transferring a part of its excitation energy to activators, which not only improves the photoluminescence of activator ions but also realizes multicolor-tunable emission in a single host. Recently, many researchers have studied the energy transfer mechanism from Ce3+ to Tb3+ or Mn2+ in some proper single host lattice for UV-based WLED application, such as KGdF4:Ce3+, Tb3+,22 CaAl2SiO6:Ce3+, Tb3+,23 La5Si2BO13:Ce3+, Mn2+,24 KBaY(BO3)2:Ce3+, Mn2+,25 and Ca3Al2O6:Ce3+, Tb3+/Mn2+.26 In these hosts, when Tb3+ or Mn2+ ions are singly doped, the luminescent properties are undesirable upon UV light excitation due to the 4f–4f weak absorption of Tb3+ and the forbidden 4T1 → 6A1 transition of Mn2+.27 By the introduction of an efficient sensitizer of Ce3+, the energy would be transferred from the 5D level of Ce3+ to the 5D3,4 level of Tb3+ or to the 4G level of Mn2+, which improve the luminescence efficiency of Tb3+ or Mn2+ ion.
Both one activator phosphors and co-doping sensitizer and activator phosphors can produce multi-color emission, however, one activator phosphors normally appeared lower CRI due to the different distribution of higher and lower energy sites for the same activator ions in the host. Considering this problem and the advantages of the co-doping sensitizer and activator phosphors, we believe that the co-doping sensitizer and activator phosphors are the most reliable and economical way to achieve multi-color emission in one host.
Among manifold multi-color emission luminescent materials, silicates have been extensively studied as the satisfactory host lattices for phosphors owing to their outstanding thermal, chemical, mechanical stability and structural diversity. For instance Ca4Y6(SiO4)6O:Ln3+ (Ln = Eu, Tb),28 CdSiO3:Eu3+,29 BaZrSi3O9:Eu3+,30 Mg2SiO4:Tb3+,31 Ba5Si8O21:Eu2+, Dy3+,32 CaAlSiN3:Ce3+,33 and so on have been reported recently. As a member of the silicate family, the cyclosilicates Sr3R2(Si3O9)2 (R = Y, Eu–Lu), has been synthesized via a solid-state reaction by Alexander P. Tyutyunnik.34 The synthesized Sr3R2(Si3O9)2 compounds corresponds to the formula of Ca3Y2(Si3O9)2, which derived from a wadeite α-CaSiO3 (pseudowollastonite, a high-temperature polymorph of CaSiO3).35,36 The Sr3R2(Si3O9)2 oxides crystallize in the monoclinic crystal system (S.G. C2/c, Z = 4) and have a morphotropic phase transition between Er and Tm compounds followed by a step-like change of the unit cell constants. There are three Sr/R sites coordinated by 8, 7 and 6 oxygen atoms for the doped ions to occupy which attracting much attention. Tyutyunnik also investigated the photoluminescence properties and applications in UV-LEDs of Sr3Y2(Si3O9)2:Eu3+ phosphor. It showed superior luminescence property in the orange-red spectral region with multiband photoluminescence emission under UV excitation, which was efficient to generate white light emitting. As one of the new cyclosilicates Sr3R2(Si3O9)2 (R = Y, Eu–Lu), Sr3Gd2(Si3O9)2 (SGSO) host material has been determined. However, to the best of our knowledge, there is no report concentrating on the luminescent properties of co-doped with multiple rare earth ions (Ce3+, Tb3+/Mn2+) and the energy transfer between them in the host of SGSO. In this study, we firstly investigated the luminescence properties, energy transfer, color tunability and thermal stability of a full-color emitting phosphor. The results reveal that the as-prepared SGSO:Ce3+, Tb3+/Mn2+ phosphors exhibit intense adjustable blue/green/white emission with high quantum yields (QYs) and thermal stability, and the energy transfer from Ce3+ to Tb3+ or Mn2+ with a high efficiency is found and studied in details.
2. Experimental
2.1 Materials and synthesis
A series of polycrystalline Sr3−zGd2−x−y(Si3O9)2:xCe3+, yTb3+/zMn2+ powder samples were prepared via conventional high temperature solid state reaction process. On the basis of the similar effective ionic radius and valence of the cations, we suggested that Ce3+/Tb3+ and Mn2+ ions prefer to occupy Gd3+ and Sr2+ sites in the host of SGSO, respectively. In order to facilitate the expression, in the following sections Sr3−zGd2−x−y(Si3O9)2:xCe3+, yTb3+/zMn2+ were abbreviated as SGSO:xCe3+, yTb3+/zMn2+ (x/y/z represent mol% in this article). The starting materials, SrCO3 (A.R.), Gd2O3 (A.R.), SiO2 (A.R.), H3BO3 (A.R.), Tb4O7 (≥99.99%) and CeO2 (≥99.99%) were thoroughly ground and mixed for 30 minutes with stoichiometric molar ratios in an agate mortar to form a homogeneous mixture. Then the mixtures were transferred into alumina crucibles and sintered at 1300 °C for 4 h under a reducing atmosphere of 95% N2–5% H2 in a horizontal tube furnace. After the furnace slowly cooled to room temperature, the calcined products were ground again, yielding the final phosphor powders.
2.2 Characterization
The phase purity of the as-prepared samples was characterized by X-ray powder diffractometer (XRD) (Bruker D8 Focus, Bruker, Kalsruhe, Germany) with Ni-filtered Cu-Kα (λ = 1.540598 Å) radiation at 40 kV tube voltage and 40 mA tube current. The XRD data were collected in a 2θ range from 5° to 70°, with the continuous scan mode at the speed of 0.05 s per step with step size of 0.01°. The measurements of photoluminescence emission and photoluminescence excitation spectra were performed by using fluorescence spectrometer (Fluoromax-4P, Horiba Jobin Yvon, New Jersey, USA) equipped with a 450 W xenon lamp as the excitation source and both excitation and emission spectra were set up to be 1.0 nm with the width of the monochromator slits adjusted as 0.50 nm. The photoluminescence decay curves were obtained from a Lecroy Wave Runner 6100 Digital Oscilloscope (1 GHz) using a tunable laser (pulse width = 4 ns, gate = 50 ns) as the excitation (Continuum Sunlite OPO). The QYs were measured by absolute photoluminescence quantum yield measurement system C9920-02.
3. Results and discussion
3.1 Phase identification, XRD refinement and crystal structure
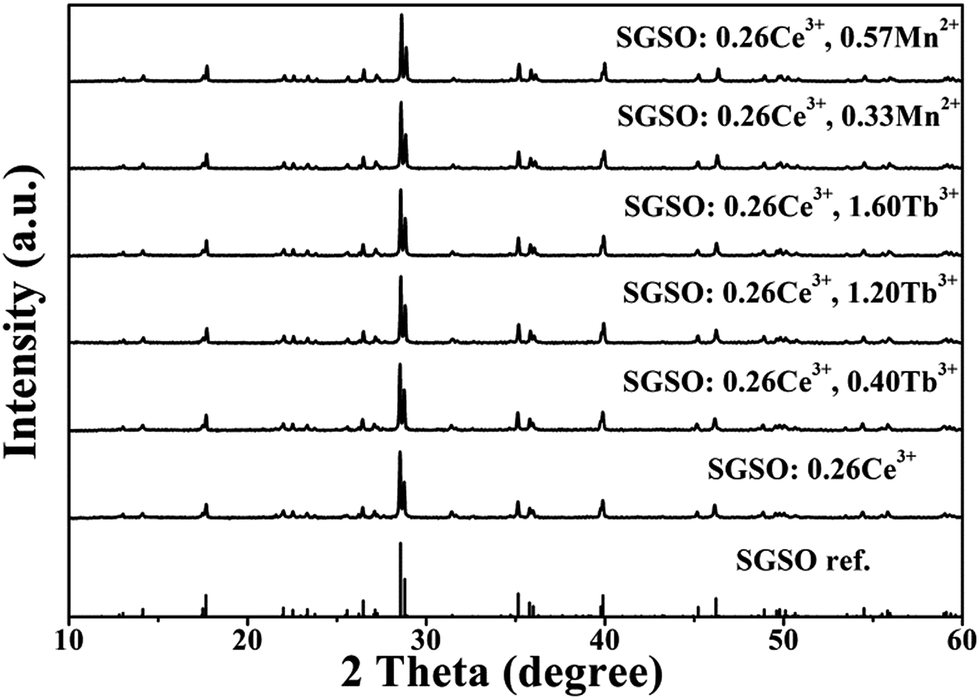
The phase purity and crystal structure of the as-prepared samples were firstly identified by XRD. Fig. 1 shows the XRD patterns of the as-prepared SGSO:0.26Ce3+, 0.40Tb3+, SGSO:0.26Ce3+, 1.20Tb3+, SGSO:0.26Ce3+, 1.60Tb3+, SGSO:0.26Ce3+, 0.33Mn2+, SGSO:0.26Ce3+, 0.57Mn2+ and the SGSO phase reported by Tyutyunnik34 as a reference. All the diffraction peaks of the samples were well indexed to the reported Sr3Gd2(Si3O9)2 phase, indicating that the obtained samples were single phase and the doped ions were completely dissolved in the SGSO host without inducing significant changes of the crystal structure. In addition, Fig. S1 (ESI†) presents the powder XRD patterns of SGSO:xCe3+ samples with the doping concentration of the Ce3+ ion ranging from 0.08 to 0.44. The patterns showed that no impurity phase can be detected and the studied samples were all pure SGSO phase. Fig. S1(b)† presents partial enlarged details of XRD patterns of SGSO:xCe3+ (x = 0.08, 0.14, 0.20, 0.26, 0.32, 0.38, 0.44). An obvious red shifting of the major diffraction peak at 2θ = 28.8° could be observed with the increase of the Ce3+ concentration.
 |
| | Fig. 1 XRD patterns of SGSO:0.26Ce3+, SGSO:0.26Ce3+, 0.40Tb3+, SGSO:0.26Ce3+, 1.20Tb3+, SGSO:0.26Ce3+, 1.60Tb3+, SGSO:0.26Ce3+, 0.33Mn2+, SGSO:0.26Ce3+, 0.57Mn2+ and the SGSO phase reported by A. P. Tyutyunnik as a reference.34 | |
In order to further understand the microstructure of the as-prepared samples, detailed Rietveld refinements were performed on the samples of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.33Mn2+. The starting model was built with crystallographic data taken from Tyutyunnik et al.34 Fig. 2 presents the results of the experimental, calculated, different XRD powder and Bragg positions for the Rietveld refinement of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.33Mn2+ at room temperature. Table 1 summarizes the final refined structural parameters, reliability factors which were from refined convergence and satisfied well with the reflection condition. It was found that the cell volume increased with the introduction of the Ce3+, Tb3+ or Mn2+ than the undoped34 which could be ascribed to the different ionic radii of the dopant ions compared with those of the SGSO host. All atom positions, fraction factors, occupation probability, thermal vibration parameters and selected interatomic distances of SGSO:0.26Ce3+, 0.60Tb3+ sample are showed in Tables S1 and S2.† The above results confirmed the phase purity of the as-prepared samples.
 |
| | Fig. 2 Experimental (crosses) and calculated (red solid line) XRD patterns and their difference (blue solid line) for the Rietveld fit of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.33Mn2+ XRD patterns by the Fullprof program. The short vertical lines show the positions of Bragg reflections of the calculated pattern. | |
Table 1 Final refined structure parameters of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.33Mn2+ derived from the Fullprof refinement of X-ray diffraction data
| Formula |
SGSO:0.26Ce3+, 0.60Tb3+ |
SGSO:0.26Ce3+, 0.33Mn2+ |
| Crystal style |
Monoclinic |
Monoclinic |
| Space group |
C2/c (15) |
C2/c (15) |
| a (Å) |
13.59469 |
13.59030 |
| b (Å) |
8.02849 |
8.01865 |
| c (Å) |
14.96769 |
14.96759 |
| β (°) |
89.60880 |
89.62453 |
| V (Å3) |
1633.608 |
1631.071 |
| Rwp (%) |
4.38 |
4.42 |
| Rp (%) |
3.15 |
3.17 |
| χ2 |
2.93 |
5.91 |
SGSO has a monoclinic phase with space group C2/c (15). The crystal structure of SGSO and the coordinated condition of Sr1/Gd1(8f), Sr2/Gd2(8f), Sr3/Gd3(4e) which are coordinated by 6, 7 and 8 oxygen atoms, respectively are depicted in Fig. 3. The structure of SGSO could be described as Sr/Gd atom layers and Si3O9 ring layers. Every SiO4 tetrahedron shared two O atoms with other SiO4 tetrahedra to construct a ternary Si3O9 ring connected by Sr/Gd atoms. Isolated Si3O9 rings were located in layers stacking with Sr/Gd layers along the [1 0 −1] direction. The ionic radius (Å) of Sr2+, Gd3+, Ce3+, Tb3+ and Mn2+ for the given coordination number (CN) are presented in Table S3.† On the basis of ionic radius and charge balance, the Ce3+/Tb3+ and Mn2+ occupied the Gd3+ and Sr2+ site, respectively. Moreover, owing to the larger Ce3+ ions substituting for the smaller Gd3+ ions, the substitution led to the increasing of interplanar spacing (d) then the decreasing of diffraction angle (2θ). This matched well with the above X-ray powder diffraction results illustrated in Fig. S1.† The similar phenomenon also existed in Fig. 1 of smaller Tb3+ or Mn2+ substituting for the larger Gd3+ and Sr2+ ions. It also verified the incorporation of Ce3+, Tb3+ and Mn2+ into the lattice and the formation of solid solutions according to the Vegard rule.37
 |
| | Fig. 3 Crystal structure schematic diagram of Sr3Gd2(Si3O9)2. | |
3.2 Photoluminescence properties of SGSO:Ce3+
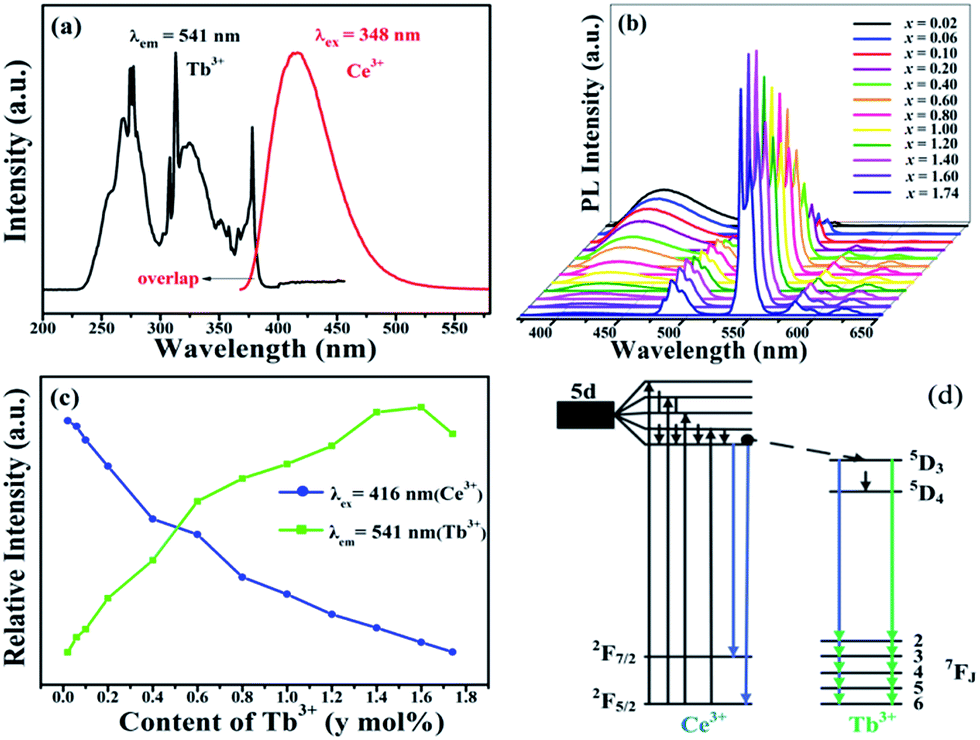
Fig. 4(a) displays the excitation and emission spectra of as obtained SGSO:0.26Ce3+ phosphors. The excitation spectrum of SGSO:0.26Ce3+ showed two broad bands centered at 295 and 348 nm (the strongest) which were attributed to the electronic transitions from the ground state of Ce3+ to the crystal field splitting of 5d states of Ce3+, as shown in Fig. 4(a). Then excited by 348 nm, the emission spectrum showed an asymmetric broad band from 370 to 500 nm with the maximum value at 416 nm, which was attributed to the 5d–4f transitions of Ce3+, implying a possible spectral overlap generated from different luminescence centers. The emission spectrum of SGSO:0.26Ce3+ was decomposed into three Gaussian components with peaks at 398, 420 and 446 nm, respectively. These components could be ascribed to that the Ce3+ ions occupied three different sites in the SGSO host which was identified with the Sr/Gd sites coordinated by different number of O atoms. It contributed to the transition from the lowest 5d excited state to the ground state of Ce3+ ions at three different Ce3+ luminescence centers. Fig. 4(b) exhibits the normalized emission spectra of SGSO:xCe3+ samples with various Ce3+ contents (x = 0.08, 0.14, 0.20, 0.26, 0.32, 0.38, 0.44). An slight red shift could be observed due to the larger Ce3+ ions substituting for the smaller Gd3+ ions as a result of the electron atmosphere of Ce3+ being compressed. The dependence of the Ce3+ luminescence intensity on its doping concentration is also displayed in inset. The highest integrated emission intensity was noted at the Ce3+ concentration of x = 0.26, which was taken as the critical concentration. The emission intensity firstly increased with adding of doping Ce3+ ions concentration, reaching the optimum point, and then decreased with doping content due to the concentration quenching effect. Concentration quenching might occur because the excitation energy migrates about a large number of centers before being emitted at the high concentrations.
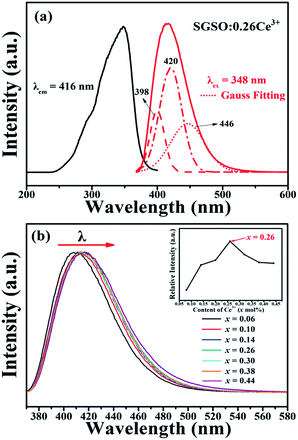 |
| | Fig. 4 (a) The excitation and emission spectra of SGSO:0.26Ce3+, together with the Gaussian peaks fitting (three red dashed lines) of the emission spectrum. (b) The normalized emission spectra of SGSO:xCe3+ (x = 0.08, 0.14, 0.20, 0.26, 0.32, 0.38, 0.44) under excitation at 348 nm. The inset is the relative intensity of Ce3+ concentration. | |
According to van Uitert, the emission position of the Ce3+ ion was strongly dependent on its local environment, which was suggested to obey an empirical relation between the energetic position of the Ce3+ emission and the local environment in diverse compounds by van Uitert38 as obeying:
| |
 | (1) |
In the above equation, E is the position for the rare-earth ion emission ion emission peak (cm−1), Q* is the position in energy for the lower d-band edge for the free ion (Q* = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 for Ce3+), V is the valance of the active ion (V = 3 for Ce3+), n is the number of anions in the immediate shell around the active ion, Ea is the electron affinity of the atoms that form anions (eV), and r is the radius of the host cation replaced by the Ce3+ion (Å). Since Ea is constant in the same host, V = 3, Q* = 50000 cm−1, the value of E is directly proportional to the product of n and r. The expected value of E determined using above equation for Ce3+ substituting GdO6 (n = 6, r = 1.05 Å), GdO7 (n = 7, r = 1.00 Å) and GdO8 (n = 8, r = 0.94 Å) was increased in turn. Therefore, we could draw the conclusion that the band centered at 398 nm was attributed to Ce3+ luminescence centers occupying the eight-coordination Sr1/Gd1 site, the band centered at 420 nm was assigned to Ce3+ ions occupying the Sr2/Gd2 site with seven-coordinated, and the band with peak at 446 nm belonged to Ce3+ luminescence centers occupying the six-coordinated Sr3/Gd3 site.
000 cm−1 for Ce3+), V is the valance of the active ion (V = 3 for Ce3+), n is the number of anions in the immediate shell around the active ion, Ea is the electron affinity of the atoms that form anions (eV), and r is the radius of the host cation replaced by the Ce3+ion (Å). Since Ea is constant in the same host, V = 3, Q* = 50000 cm−1, the value of E is directly proportional to the product of n and r. The expected value of E determined using above equation for Ce3+ substituting GdO6 (n = 6, r = 1.05 Å), GdO7 (n = 7, r = 1.00 Å) and GdO8 (n = 8, r = 0.94 Å) was increased in turn. Therefore, we could draw the conclusion that the band centered at 398 nm was attributed to Ce3+ luminescence centers occupying the eight-coordination Sr1/Gd1 site, the band centered at 420 nm was assigned to Ce3+ ions occupying the Sr2/Gd2 site with seven-coordinated, and the band with peak at 446 nm belonged to Ce3+ luminescence centers occupying the six-coordinated Sr3/Gd3 site.
3.3 Photoluminescence properties and energy transfer of SGSO:Ce3+, Tb3+
The excitation and emission spectra of the Tb3+ singly doped SGSO matrix were studied in our other work. Here, the Ce3+ → Tb3+ energy transfer in SGSO:Ce3+, Tb3+ system was investigated in detail. Fig. 5(a) demonstrates a significant overlap between the emission band of Ce3+ and the excitation band of Tb3+. According to the energy transfer formula, which was given by Dexter39 is expressed as the following equation:| |
 | (2) |
where PSA is energy transfer rate and HSA is the interaction Hamiltonian that mediates energy transfer from the excited sensitizers to the unexcited activators. The matrix element indicates the interaction between the initial state |S*,A〉 and the final state 〈S,A*|. The integral represents the spectral overlap between the emission spectrum of the sensitizers and the excitation spectrum of the activators. From the observed spectral overlap between the emission band of Ce3+ and the excitation peaks of Tb3+, we could conclude from above equation that a possible resonance type energy transfer might occur in SGSO:Ce3+, Tb3+ samples.2
 |
| | Fig. 5 (a) The spectral overlap between the emission band of Ce3+ and the excitation peaks of Tb3+ (b) the emission spectra of SGSO:0.26Ce3+, yTb3+ (y = 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) under excitation at 348 nm. (c) The emission relative intensity of Ce3+ and Tb3+ (d) the energy level diagram of Ce3+, Tb3+. | |
Fig. 5(b) shows a series of emission spectra for SGSO:0.26Ce3+, yTb3+ (y = 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) under UV excitation (λex = 348 nm). It could be revealed from Fig. 5(c) that the emission intensity of Ce3+ gradually decreased while the emission intensity of Tb3+ gradually increased with the increase of Tb3+ concentration, making further confirmations on the existence of Ce3+ → Tb3+ energy transfer in SGSO:Ce3+, Tb3+. The Tb3+ emission intensity was saturated when the Tb3+ dopant content (y) was above 1.60. This was attributed to the concentration quenching effect, which occurred when the activator concentration was high. To comprehend the process of energy transfer in the co-doped SGSO phosphors more clearly, the energy level diagram is given by the inset in Fig. 5(d). In SGSO:Ce3+, Tb3+ phosphor with exciting Ce3+, electrons were excited from the ground state, 2F5/2, to the 5d excited state of Ce3+ simultaneously due to the level of 5d excited state of Ce3+ was close to the level of 5D4 of Tb3+, there was a certain probability that the energy transfer from 5d excited state of Ce3+ to 5D4 of Tb3+ through relaxation and nonradiative progress. The transitions of 5D4–7F5 of Tb3+ led to release 541 nm photons.27
In order to further detect the Ce3+ → Tb3+ energy transfer process in the SGSO host lattice, the lifetime dependence of Ce3+ on the concentration of Tb3+ in SGSO:0.26Ce3+, yTb3+ (y = 0, 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) phosphors were measured at 348 nm excited and monitored at 416 nm, the decay curves are shown in Fig. 6. Since there were three Ce3+ luminescent centers in the SGSO host, the decay curves were successfully fitted using the following three exponential equation:40,41
| | |
I(t) = I0 + A1exp(−t/τ1) + A2exp(−t/τ2) + A3exp(−t/τ3)
| (3) |
where
I(
t) and
I0 are the luminescence intensities at times
t;
A1,
A2 and
A3 are fitting constants and
τ1,
τ2 and
τ3 are the decay times for the exponential components. Using these parameters, the average decay times (
τ*) can be determined by the formula as follows:
42| |
 | (4) |
 |
| | Fig. 6 Decay curves of Ce3+ emission monitored at 416 nm for SGSO:0.26Ce3+, yTb3+ (y = 0, 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) under excitation at 348 nm. The inset shows the dependence of energy transfer efficiency (ηT) on the Tb3+ content. | |
The average decay times τ were calculated to be 34.9, 31.7, 29.6, 25.6, 21.6, 19.3, 17.4, 14.8, 14.3 and 14.1 ns for SGSO:0.26Ce3+, yTb3+ with y = 0, 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60 and 1.74, respectively.
The energy transfer efficiency (ηT) could be calculated using the following equation by Paulose et al.43
| |
 | (5) |
where
τS0 and
τS stand for the lifetimes of Ce
3+ in the absence and the presence of Tb
3+, respectively. Results showed that the
ηT values from Ce
3+ to Tb
3+ for SGSO:0.26Ce
3+,
yTb
3+ (
y = 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) were calculated to be 9.2%, 15.2%, 26.6%, 38.1%, 44.7%, 50.1%, 57.6%, 59.0%, 59.6% respectively. The inset of
Fig. 6 shows the dependence of energy transfer efficiency (
ηT) on the Tb
3+ content. All results above proved that the energy transfer process from Ce
3+ to Tb
3+ in the SGSO host is very efficient.
In order to understand the energy transfer mechanism of SGSO:Ce3+, Tb3+, it was available to know the critical distance RC, which is defined as the distance for which the probability of transfer equals to the probability of radiative emission of energy donator by the ion of Ce3+ in this system.44 In this passage, the RC was first calculated by using the concentration quenching method, where the critical distance RC between Ce3+ and Tb3+ could be estimated by the following formula given by Blasse.39
| |
 | (6) |
where
V is the volume of the unit cell, and
N is the number of cations in the unit cell.
XC is the critical concentration at which the luminescence intensity of Ce
3+ as a sensitizer was half of that in the sample in the absence of Tb
3+ as an activator, that is to say,
XC occurred when
ηT = 0.5. In this case,
N = 4,
V = 1633.608 Å
3 and
XC was 0.60 in the SGSO:Ce
3+, Tb
3+ system. Accordingly, the critical distance
RC was calculated to be about 10.91 Å for Ce
3+ and Tb
3+ in SGSO host.
Exchange interactions and multipolar interactions are the two main aspects responsible for the resonant energy transfer mechanism. In general, if energy transfer results from the exchange interactions, the critical distance (RC) between the sensitizer and activator should be shorter than 4 Å. In this system, the critical distance of energy transfer (RC) was relatively long, the exchange interaction could be disregarded.45 In accordance with Dexter's energy transfer expressions of exchange and multipolar interaction and Reisfeld's approximation, the following relationship could be formed as:39,46
where
y is the concentration of Tb
3+,
IS0 and
IS are the luminescence intensity of a sensitizer (Ce
3+) in the absence and presence of an activator (Tb
3+), respectively.
IS0/
IS ∝
yn/3, with
n = 6, 8, and 10 corresponds to dipole–dipole (d–d), dipole–quadrupole (d–q) and quadrupole–quadrupole (q–q) interactions, respectively.
47 The relationships between
IS0/
IS with
yn/3 are illustrated in
Fig. 7(a)–(c). The linear relationship reached the optimal one for
IS0/
IS ∝
yn/3 by comparing the fitting factors of
R values in
Fig. 7(b), implying that energy transfer from Ce
3+ to Tb
3+ occurred
via the dipole–quadrupole interactions.
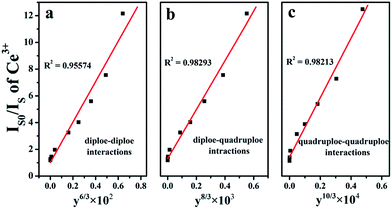 |
| | Fig. 7 Dependence of IS0/IS of Ce3+ in SGSO:0.26Ce3+, yTb3+ (y = 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) samples, (a) y6/3, (b) y8/3 and (c) y10/3. | |
Considering the dipole–quadrupole interaction, the critical distance from the sensitizer to the activator could also be calculated by the spectral overlap method, as simplified expression by:26
| |
 | (8) |
where
fq is the oscillator strength of the electric quadrupole transition of the activator ion (Tb
3+),
λS (Å) is the wavelength corresponding to the strongest emission peak of the sensitizer, and
E is the energy (eV) corresponding to the largest emission wavelength. ∫
FS(
E)
FA(
E)d
E/
E4 represents the spectral overlap between the normalized emission spectrum,
FS(
E), of the Ce
3+ and the excitation spectrum,
FA(
E), of Tb
3+. However, the oscillator strength of the Tb
3+ quadrupole transition (
fq) did not obtained up to now. It was suggested by Verstegen
et al. that the ratio
fq/
fd is about 1 × 10
−2 to 1 × 10
−3 when
fd applies to a forbidden dipole transition.
48 fd is the oscillator strength of the involved dipole absorption transition of the activator. In our case, using above equation with
λS = 4140 Å,
fq = 10
−2 to 10
−3 fd,
fd of the Tb
3+ transition is 0.3 × 10
−6, ∫
FS(
E)
FA(
E)d
E/
E4 was calculated to be about 0.01211 eV
−5, and the critical distance (
RC) was calculated to be 10.82–14.43 Å for the dipole–quadrupole interaction. This agreed approximately with the concentration-quenching method (10.91 Å), which further concluded that the mechanism of energy transfer from the Ce
3+ to Tb
3+ ions was mainly due to a dipole–quadrupole interaction.
3.4 Photoluminescence properties and energy transfer of SGSO:Ce3+, Mn2+
It is known that Mn2+ generally shows a broad emission band because of the 4T1–6A1 transition within the 3d shell, in which the electrons are strongly coupled to lattice vibrations and are affected by the crystal field strength and site symmetry.49 The emission color of Mn2+ can vary from green under strong crystal field to orange/red under weak crystal field. However, the excitation transitions of Mn2+ are difficult to pump and the emission intensity is very weak due to the forbidden d–d transitions. In order to enhance the absorption and emission of Mn2+, we designed the Ce3+ → Mn2+ energy transfer in SGSO host and studied the luminescence properties and energy transfer of SGSO:Ce3+, Mn2+ samples in detail. As shown in Fig. 8, the emission band of Ce3+ and the excitation peaks of Mn2+ were presented. The excitation peaks of Mn2+ consisted of two parts: one part included two sharp peaks at 274 and 313 nm, corresponding to the 8S → 6I and 8S → 6P transitions of Gd3+ ions in the host, respectively;50 and the other part contained several peaks located at 344, 361, 406, 449, and 468 nm corresponding to the transitions of Mn2+ from 6A1(6S) to 4E(4D), 4T2(4D), [4A1(4G), 4E(4G)], 4T2(4G), and 4T1(4G), respectively. Moreover, from Fig. 8 we could also find that there existed a significant overlap between the excitation spectrum of SGSO:Mn2+ (370–460 nm) and emission spectrum of SGSO:Ce3+. As a consequence, there likely had a strong resonant energy transfer between the sensitizer Ce3+ and the activator Mn2+.
 |
| | Fig. 8 The excitation spectrum of SGSO:Mn2+ and the emission spectrum of SGSO:Ce3+. | |
Inspection of Fig. 9(a) shows the emission spectra of SGSO:0.26Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) under 348 nm excitation, which showed not only a broad blue band peaked at 416 nm due to the transitions of 5d → 4f of Ce3+ but also a green-emitting band (500–650 nm, 4T1 → 6A1) of the Mn2+ ions. With increasing Mn2+ concentration, the emission intensities of Ce3+ decreased by degrees, whereas the emission intensities of Mn2+ increased at first then decreased with quenching concentration at z = 0.57 (Fig. 9(b)), further reflecting the result of energy transfer from Ce3+ to Mn2+.
 |
| | Fig. 9 (a) The emission spectra of SGSO:0.26Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) under excitation at 348 nm. (b) The emission relative intensity of Ce3+ and Mn2+. | |
Similarly, for SGSO:0.26Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81), the lifetime of Ce3+ decreased with increasing Mn2+ concentration, as 34.9, 33.8, 32.9, 31.9, 30.9, 30.0, 28.5 ns, respectively. Fig. S2† shows that the luminescence lifetime of Ce3+ decreases with increasing Mn2+ concentration because the energy transfer from Ce3+ to Mn2+.
The energy transfer mechanism between the sensitizer (Ce3+) and activator (Mn2+) was also calculated with eqn (7). Fig. S3† shows the relationship between IS0/IS and zn/3 in SGSO:0.26Ce3+, zMn2+ when n = 6, 8, 10. The curve for IS0/IS vs. zn/3 was closer to a linear relationship when n = 6, and this clearly indicated that the energy transfer between Ce3+ and Mn2+ was the dipole–dipole mechanism in the SGSO host. The critical distances (RC) calculated by the quenching concentration method was 11.10 Å. All of these results indicated the efficient energy transfer from Ce3+ to Mn2+.
3.5 Thermal photoluminescence properties of SGSO:Ce3+, Tb3+/Mn2+
The thermal quenching of phosphors applied in devices is one of the most important technological parameters. The temperature of the devices usually increases to 100–150 °C and even higher, which related to the chromaticity and brightness of phosphors.2 Consequently, the thermal photoluminescence stability of devices depends strongly on phosphors. Thus, the temperature dependence emission spectra of SGSO:0.26Ce3+ under 348 nm in the temperature range of 25 °C to 200 °C were shown in Fig. 10. It could be seen that the luminescence intensity of Ce3+ had an obvious decreasing tendency with increasing temperature, and the luminescence intensity dropped to 62.6% of the initial intensity when the temperature was raised up to 150 °C. The temperature dependent luminescence behavior of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.45Mn2+ under 348 nm excitation are also depicted in Fig. 11. The temperature dependence of the integral emissions of Ce3+, Tb3+ and Mn2+ are also illustrated, respectively. Both the integral intensity of Tb3+ emission and that of Mn2+ decreased more gradually than Ce3+ with increasing temperature. The luminescence intensity of Tb3+ and Mn2+ decreased to 72.3% and 86.1% at 150 °C comparing to room temperature. These phenomena provided the proof that the energy transfer efficiency through Ce3+ → Tb3+ and Ce3+ → Mn2+ was observably influenced by temperature. When Ce3+ ion was irradiated by 348 nm light excitation, electron was pumped to the higher component of 5d level, and then relaxed to the lower excited state (5D3/2, ∼22988) of Ce3+ ion, and the excited state (5D4) of Tb3+ ion was at ∼20576 cm−1. The difference between the two close levels was about 2412 cm−1. It was reported that the vibration frequency for a perfectly symmetric SiO4 tetrahedron was about 608 cm−1.51 Therefore, it was reasonable and probably that the energy difference between the lowest excited state of Ce3+ ion and the excited state (5D4) of Tb3+ ion was bridged by the activated phonons in SGSO host. In conclusion, there proposed thermally activated phonon-assisted energy transfer through Ce3+ → Tb3+ in SGSO.52,53 The same principle could explain the increasing integral intensity ratio of Mn2+/Ce3+ with temperature increasing.
 |
| | Fig. 10 Temperature-dependent emission spectra of SGSO:0.26Ce3+ under different temperatures in the range of 25–200 °C. | |
 |
| | Fig. 11 Temperature-dependent emission spectra of (a) SGSO:0.26Ce3+, 0.60Tb3+, (b) SGSO:0.26Ce3+, 0.45Mn2+. The variations of the relative emission intensities of (c) SGSO:0.26Ce3+, 0.60Tb3+, (d) SGSO:0.26Ce3+, 0.45Mn2+. | |
To better understand the temperature dependence of photoluminescence, the activation energy (ΔE was calculated. The activation energy for thermal quenching was estimated using the Arrhenius equation:54,55
| |
 | (9) |
where
I0 is the initial emission intensity of the phosphor at room temperature,
IT is the luminous intensity at different temperatures,
c is a constant, Δ
E is the activation energy for thermal quenching, and
k is Boltzmann constant (8.62 × 10
−5 eV). According to the equation, the activation energy Δ
E could be calculated from a plotting of ln[(
I0/
IT) − 1] against 1/
kT, where a straight slope equals −Δ
E. As shown in
Fig. 12, Δ
E was found to be 0.2013 and 0.1233 eV for Tb
3+ and Mn
2+, respectively. In addition, the Δ
E for single doped of Ce
3+ as a sensitizer was also investigated. When Ce
3+ was single doped in SGSO host, the Δ
E was calculated to be 0.2560 eV. However, the Δ
E of Ce
3+ increased to 0.3048 eV and 0.2887 eV as co-doping Tb
3+ and Mn
2+. The relatively high activation energy resulted in a good thermal stability for this phosphor. This phenomenon might be ascribed to the weakness of the 5d orbits splitting of Ce
3+. When smaller Tb
3+ or Mn
2+ ions substituted the larger Gd/Sr sites, the crystal structure became looser, resulting in the slackness of crystal field strength around Ce
3+. To express the variation of Δ
E of Ce
3+ more vividly, the configurational coordinate diagram of the ground state of Ce
3+ and the excited states of Ce
3+ is exhibited in
Fig. 13. Under the 348 nm light excitation, the electrons were excited to the excited states from the ground to
5D
3/2. At the room temperature, most of the electrons returned to the ground state along the way ①, which emitted light. But with the increased of temperature, most electrons absorbing activation energy Δ
E returned to the ground along the way ②, which was the non-radiative transition. Here, the energy difference between
5D
3/2 and
5D
5/2 became narrower due to the change of crystal field strength around Ce
3+. As a consequence, after absorbing activation energy Δ
E, most electrons returned to the ground along the way ③. The co-doping of Tb
3+ or Mn
2+ provided a hard way for electrons returning to the ground state, which was the reason for the higher thermal stability of Ce
3+. Additionally, with the integral intensity of Tb
3+ or Mn
2+ decreased more gradually than Ce
3+ by increasing temperature, the CIE chromaticity coordinates shifted to green region with increasing temperature, as shown in
Fig. 14. However, the emission color still located in a small region from 25 °C to 200 °C, as given in the dashed red circles in the CIE diagram. The same phenomenon was found from the previous report
ref. 47.
 |
| | Fig. 12 ln[(I0/IT) − 1] versus 1/kT plot and the thermal activation energy of SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.45Mn2+. | |
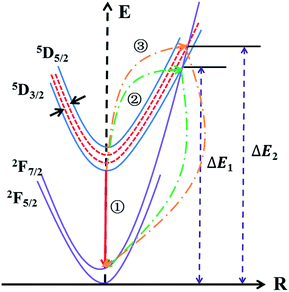 |
| | Fig. 13 The configurational coordinate diagram of the ground states and the excited states of Ce3+. | |
 |
| | Fig. 14 The CIE chromaticity coordinates as a function of temperature of the SGSO:0.26Ce3+, 0.60Tb3+ and SGSO:0.26Ce3+, 0.45Mn2+. | |
3.6 QYs and CIE of SGSO:Ce3+, Tb3+/Mn2+
QYs is defined as the ratio of the number of photons emitted to the number of photons absorbed in unit time. For solid state lighting applications, the QY is an important parameter to be considered. To accurately investigate the luminescence properties of phosphors, the QYs of SGSO:Ce3+, Tb3+/Mn2+ phosphors under 348 nm UV excitation were measured and listed in Table 2. It could be seen that the QYs of Ce3+ single doped SGSO phosphor was 60.2%. However, when Tb3+ or Mn2+ was co-doped with Ce3+ in the SGSO host, the QYs of SGSO:Ce3+, Tb3+/Mn2+ decreased firstly with the increase of Tb3+ or Mn2+ content which indicating the Ce3+ → Tb3+ and Ce3+ → Mn2+ energy transferred happen in SGSO host. The maximum QYs of as-prepared SGSO:Ce3+, Tb3+ samples could reach 80.2% at y = 0.10, which demonstrated that efficient energy transfer could occur between Ce3+ and Tb3+ ions. Moreover, by optimizing the experimental conditions and the compositions of the phosphors, the QYs could be improved. The energy transfer from sensitizer to activator was a viable route to realize color-tunable emission in single host, and a white light emission could be obtained through adjusting the concentration of sensitizer and activator at a suitable ratio. In this SGSO:Ce3+, yTb3+/zMn2+ system, it was hopeful to realize color-tunable emission even white light emission through energy transfer due to the blue and green emission of Ce3+, Tb3+ and Mn2+. Thereby, the CIE chromaticity coordinate of SGSO:Ce3+, yTb3+/zMn2+ (y = 0, 0.02, 0.06, 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) phosphors under 348 nm excitation which were calculated based on the corresponding emission spectra is showed in Fig. 15. From the representing features of chromaticity coordinates above, we could find that the color tone of the SGSO:Ce3+, yTb3+ (y = 0, 0.02, 0.06, 0.10, 0.20, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) phosphors shifted gradually from blue to green position with the increasing of Tb3+ concentration. The color hue of SGSO:Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) phosphors changed from blue to near white region with the increase of z values. In order to have a more intuitive expression for the emission colors of our prepared samples, the digital photographs taken under a 365 nm UV lamp are also shown in Fig. 15. Based on these results, the obtained phosphors could act as potential color tunable phosphors for its possible applications in solid-state lighting and display.
Table 2 The CIE chromaticity coordinates (x, y) and QYs of SGSO:0.26Ce3+, yTb3+ (y = 0, 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) and SGSO:Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81)
| No. |
Composition |
CIE (x, y) |
QYs (%) |
| 0 |
SGSO:0.26Ce3+ |
0.174, 0.060 |
60.2 |
| 1 |
SGSO:0.26Ce3+, 0.02Tb3+ |
0.174, 0.060 |
25.8 |
| 2 |
SGSO:0.26Ce3+, 0.06Tb3+ |
0.193, 0.173 |
33.2 |
| 3 |
SGSO:0.26Ce3+, 0.10Tb3+ |
0.199, 0.205 |
80.2 |
| 4 |
SGSO:0.26Ce3+, 0.20Tb3+ |
0.222, 0.318 |
68.6 |
| 5 |
SGSO:0.26Ce3+, 0.40Tb3+ |
0.232, 0.363 |
62.8 |
| 6 |
SGSO:0.26Ce3+, 0.60Tb3+ |
0.263, 0.510 |
47.7 |
| 7 |
SGSO:0.26Ce3+, 0.80Tb3+ |
0.272, 0.550 |
50.2 |
| 8 |
SGSO:0.26Ce3+, 1.00Tb3+ |
0.276, 0.571 |
52.1 |
| 9 |
SGSO:0.26Ce3+, 1.20Tb3+ |
0.280, 0.590 |
76.3 |
| 10 |
SGSO:0.26Ce3+, 1.40Tb3+ |
0.282, 0.596 |
59.6 |
| 11 |
SGSO:0.26Ce3+, 1.60Tb3+ |
0.284, 0.609 |
54.8 |
| 12 |
SGSO:0.26Ce3+, 1.74Tb3+ |
0.286, 0.615 |
56.6 |
| 13 |
SGSO:0.26Ce3+, 0.09Mn2+ |
0.178, 0.076 |
55.2 |
| 14 |
SGSO:0.26Ce3+, 0.21Mn2+ |
0.198, 0.123 |
44.0 |
| 15 |
SGSO:0.26Ce3+, 0.33Mn2+ |
0.222, 0.195 |
61.3 |
| 16 |
SGSO:0.26Ce3+, 0.45Mn2+ |
0.241, 0.236 |
60.8 |
| 17 |
SGSO:0.26Ce3+, 0.57Mn2+ |
0.263, 0.280 |
62.4 |
| 18 |
SGSO:0.26Ce3+, 0.69Mn2+ |
0.272, 0.296 |
51.5 |
| 19 |
SGSO:0.26Ce3+, 0.81Mn2+ |
0.287, 0.323 |
42.1 |
| 20 |
SGSO:0.26Ce3+, 0.93Mn2+ |
0.306, 0.347 |
— |
| 21 |
SGSO:0.26Ce3+, 1.05Mn2+ |
0.319, 0.367 |
— |
 |
| | Fig. 15 The CIE chromaticity coordinates diagram for SGSO:0.26Ce3+, yTb3+ (y = 0, 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) and SGSO:Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81). | |
4. Conclusion
In summary, we have successfully developed novel bluish-green-white tunable emitting SGSO:Ce3+, Tb3+/Mn2+ phosphors by the conventional solid-state reaction. For the samples doped with only Ce3+ ions, an asymmetric emission spectrum peaking at 416 nm was observed due to the 4f–5d transition of Ce3+, and the optimal concentration was 0.26. When Ce3+ and Tb3+/Mn2+ ions were co-doped into the host, the spectroscopic data of SGSO:Ce3+, SGSO:0.26Ce3+, yTb3+ (y = 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) and SGSO:Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) indicated that the Ce3+ to Tb3+ or Mn2+ energy transfer process occurred in the SGSO host under near-UV excitation. The Ce3+ to Tb3+ or Mn2+ energy transfer in SGSO phosphors were demonstrated to be a resonant type via the dipole–quadrupole mechanism and dipole–dipole mechanism. Due to the energy transfer from Ce3+ → Tb3+ and Ce3+ → Mn2+, both the integral intensity of Tb3+ emission and that of Mn2+ decreased more gradually than Ce3+ with increasing temperature. The luminescence intensity of Tb3+ and Mn2+ decreased to 72.3% and 86.1% at 150 °C comparing to room temperature. Compared to single doped Ce3+, the co-doping of Tb3+ or Mn2+ resulted in the higher thermal stability of Ce3+ because Tb3+ or Mn2+ ions provided a hard way for electrons returning to the ground state of Ce3+. And the QYs of the SGSO:0.26Ce3+, 0.10Tb3+ and SGSO:0.26Ce3+, 0.57Mn2+ phosphors reached 80.2% and 62.4% under the excitation of 348 nm. The emission color of SGSO:0.26Ce3+, yTb3+ (y = 0.02, 0.06, 0.10, 0.20, 0.40, 0.60, 0.80, 1.00, 1.20, 1.40, 1.60, 1.74) and SGSO:Ce3+, zMn2+ (z = 0, 0.09, 0.21, 0.33, 0.45, 0.57, 0.69, 0.81) phosphors tuned from deep blue to green and white via controlling the concentration of Tb3+ or Mn2+. These results indicate that these novel phosphors would be single-phase color-tunable phosphors for ultraviolet-convertible devices.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21571162, 21171152, 21301162), the Guangdong Province Enterprise-University-Academy Collaborative Project (No. 2012B091100474) and Open Foundation of Hubei key laboratory of low dimensional optoelectronic materials and devices (No. HLOM142002).
References
- X. F. Li, J. D. Budai, F. Liu, J. Y. Howe, J. H. Zhang, X. J. Wang, Z. J. Gu, C. J. Sun, R. S. Meltzer and Z. W. Pan, Light: Sci. Appl., 2013, 2, 1 Search PubMed.
- H. K. Liu, Y. Luo, Z. Y. Mao, L. B. Liao and Z. G. Xia, J. Mater. Chem. C, 2014, 2, 1619 RSC.
- C. H. Huang, T. M. Chen, W. R. Liu, Y. C. Chiu, Y. T. Yeh and S. M. Jang, ACS Appl. Mater. Interfaces, 2010, 2, 259 CAS.
- N. Guo, Y. J. Huang, H. P. You, M. Yang, Y. H. Song, K. Liu and Y. H. Zheng, Inorg. Chem., 2010, 49, 10907 CrossRef CAS PubMed.
- J. S. Hou, W. Z. Jiang, Y. Z. Fang and F. Q. Huang, J. Mater. Chem. C, 2013, 1, 5892 RSC.
- T. Pulli, T. Dönsberg, T. Poikonen, F. Manoocheri, P. Kärhä and E. Ikonen, Light: Sci. Appl., 2015, 4, 332 CrossRef.
- Z. G. Xia, C. G. Ma, M. S. Molokeev, Q. L. Liu, K. Rickert and K. R. Poeppelmeier, J. Am. Chem. Soc., 2015, 137, 12494 CrossRef CAS PubMed.
- J. Zhou, Z. G. Xia, M. Y. Chen, M. S. Molokeev and Q. L. Liu, Sci. Rep., 2015, 5, 12149 CrossRef PubMed.
- A. A. Setlur, W. J. Heward, M. E. Hannah and U. Happek, Chem. Mater., 2008, 20, 6277 CrossRef CAS.
- Y. F. Liu, X. Zhang, Z. D. Hao, X. J. Wang and J. H. Zhang, J. Mater. Chem., 2011, 21, 6354 RSC.
- X. F. Zhou, Z. Y. Zhang and Y. H. Wang, J. Mater. Chem. C, 2015, 3, 3676 RSC.
- Q. F. Guo, L. B. Liao and Z. G. Xia, J. Lumin., 2014, 145, 65 CrossRef CAS.
- Y. M. Feng, J. P. Huang, L. L. Liu, J. Liu and X. B. Yu, Dalton Trans., 2015, 44, 15006 RSC.
- W. Z. Lv, N. Guo, Y. C. Jia, Q. Zhao and H. P. You, Opt. Mater., 2013, 35, 1013 CrossRef CAS.
- J. Zhou, Z. G. Xia, H. P. You, K. Shen, M. X. Yang and L. B. Liao, J. Lumin., 2013, 135, 20 CrossRef CAS.
- B. H. Li, J. Yang, J. Wang and M. M. Wu, Opt. Mater., 2014, 36, 1649 CrossRef CAS.
- M. F. Zhang, Y. J. Liang, Z. G. Xia, F. Yang, D. Y. Yu and G. G. Li, Mater. Res. Bull., 2013, 48, 4749 CrossRef CAS.
- Z. G. Xia and W. W. Wu, Dalton Trans., 2013, 42, 12989 RSC.
- Z. P. Ci, Q. S. Sun, M. X. Sun, X. J. Jiang, S. C. Qin and Y. H. Wang, J. Mater. Chem. C, 2014, 2, 5850 RSC.
- D. X. Wei, Y. Sun, L. Z. Jiang, S. X. Hu and D. Li, New J. Chem., 2015, 39, 4753 RSC.
- Y. Y. Li, J. Y. Ding, Q. S. Wu, Q. Long, X. C. Wang and Y. H. Wang, J. Mater. Chem. C, 2015, 3, 8949 RSC.
- C. Y. Cao, H. K. Yang, J. W. Chung, B. K. Moon, B. C. Choi, J. H. Jeong and K. H. Kim, J. Mater. Chem., 2011, 21, 10342 RSC.
- B. L. Wang, L. Z. Sun and H. D. Ju, Solid State Commun., 2010, 150, 1460 CrossRef CAS.
- H. K. Liu, L. B. Liao and Z. G. Xia, RSC Adv., 2014, 4, 7288 RSC.
- Z. P. Lian, J. F. Sun, L. J. Zhang, D. Z. Shen, G. Q. Shen, X. Q. Wang and Q. F. Yan, RSC Adv., 2013, 3, 16534 RSC.
- J. L. Zhang, Y. N. He, Z. X. Qiu, W. L. Zhang, W. L. Zhou, L. P. Yu and S. X. Lian, Dalton Trans., 2014, 43, 18134 RSC.
- W. Lv, N. Guo, Y. C. Jia, Q. Zhao, W. Z. Lv, M. M. Jiao, B. Q. Shao and H. P. You, Inorg. Chem., 2013, 52, 3007 CrossRef PubMed.
- C. Peng, G. G. Li, X. J. Kang, C. X. Li and J. Lin, J. Colloid Interface Sci., 2011, 355, 89–95 CrossRef CAS PubMed.
- H. Nagabhushana, D. V. Sunitha, S. C. Sharma, S. C. Prashantha, B. M. Nagabhushana and R. P. S. Chakradhar, J. Alloys Compd., 2014, 616, 284 CrossRef CAS.
- T. Komukai, Y. Takatsuka, H. Kato and M. Kakihana, J. Lumin., 2015, 158, 328 CrossRef CAS.
- R. Naik, S. C. Prashantha, H. Nagabhushana, H. P. Nagaswarupa, K. S. Anantharaju, B. M. Nagabhushana, H. B. Premkumar and K. M. Girish, J. Alloys Compd., 2014, 617, 69 CrossRef CAS.
- P. J. Wang, X. H. Xu, D. C. Zhou, X. Yu and J. B. Qiu, Inorg. Chem., 2015, 54, 1690 CrossRef CAS PubMed.
- Y. Q. Li, N. Hirosaki, R. J. Xie, T. Takeda and M. Mitomo, Chem. Mater., 2008, 20, 6704 CrossRef CAS.
- A. P. Tyutyunnik, I. I. Leonidov, L. L. Surat, I. F. Berger and V. G. Zubkov, J. Solid State Chem., 2013, 197, 447 CrossRef CAS.
- T. Yamanaka and H. Mori, Acta Crystallogr., B, 1981, 37, 1010 CrossRef.
- H. Yamane, T. Nagasawa, M. Shimada and T. Endo, Acta Crystallogr., C, 1997, 53, 1533 Search PubMed.
- C. W. Yeh, W. T. Chen, R. S. Liu, S. F. Hu, H. S. Sheu, J. M. Chen and H. T. Hintzen, J. Am. Chem. Soc., 2012, 134, 14108 CrossRef CAS PubMed.
- L. G. van Uitert, J. Lumin., 1984, 29, 1 CrossRef CAS.
- J. Zhou and Z. G. Xia, J. Mater. Chem. C, 2015, 3, 7552 RSC.
- G. Zhu, S. Y. Xin, Y. Wen, Q. Wang, M. D. Que and Y. H. Wang, RSC Adv., 2013, 3, 9311 RSC.
- B. M. Mothudi, O. M. Ntwaeaborwa, S. S. Pitale and H. C. Swart, J. Alloys Compd., 2010, 508, 262 CrossRef CAS.
- M. F. Zhang, Y. J. Liang, R. Tang, D. Y. Yu, M. H. Tong, Q. Wang, Y. L. Zhu, X. Y. Wu and G. G. Li, RSC Adv., 2014, 4, 40626 RSC.
- F. Y. Xie, J. H. Li, Z. Y. Dong, D. W. Wen, J. X. Shi, J. Yan and M. M. Wu, RSC Adv., 2015, 5, 59830 RSC.
- Y. Wen, Y. H. Wang, F. Zhang and B. T. Liu, Mater. Chem. Phys., 2011, 129, 1171 CrossRef CAS.
- R. Reisfeld, E. Greenberg, R. Velapoldi and B. Barnett, J. Chem. Phys., 1972, 56, 1698 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- J. Zhou and Z. G. Xia, J. Mater. Chem. C, 2014, 2, 6978 RSC.
- J. M. P. J. Verstegen, J. L. Sommerdijk and J. G. Verriet, J. Lumin., 1973, 6, 425 CrossRef CAS.
- L. Shi, Y. L. Huang and H. J. Seo, J. Phys. Chem. A, 2010, 114, 6927 CrossRef CAS PubMed.
- J. He, S. Zhang, J. B. Zhou, J. P. Zhong, H. B. Liang, S. S. Sun, Y. Huang and Y. Tao, Opt. Mater., 2015, 39, 81 CrossRef CAS.
- R. L. Frost, A. López, Y. F. Xi and R. Scholz, Spectrochim. Acta, Part A, 2015, 137, 717 CrossRef CAS PubMed.
- J. Y. Sun, J. C. Zhu, X. Y. Zhang, Z. G. Xia and H. Y. Du, J. Lumin., 2012, 132, 2937 CrossRef CAS.
- H. D. Luo, J. Liu, X. Zheng, L. X. Han, K. X. Ren and X. B. Yu, J. Mater. Chem., 2012, 7, 15892 Search PubMed.
- J. S. Kim, H. J. Song, H. S. Roh, D. K. Yim, J. H. Noh and K. S. Hong, Mater. Lett., 2012, 79, 112 CrossRef CAS.
- J. Zhou and Z. G. Xia, J. Lumin., 2014, 146, 22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20756h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.