
BSA blocking in enzyme-linked immunosorbent assays is a non-mandatory step: a perspective study on mechanism of BSA blocking in common ELISA protocols†
Rajesh Ahirwarab,
Shilpi Bariara,
Abitha Balakrishnana and
Pradip Nahar*ab
aCSIR-Institute of Genomics and Integrative Biology, Mall Road Delhi, Delhi, India-110007. E-mail: pnahar@igib.res.in
bAcademy of Scientific and Innovative Research, Delhi, 110007, India
First published on 10th November 2015
Abstract
BSA blocking is a routine practice among clinicians and researchers working on immunoassays throughout the world. The primary role of BSA is to prevent the non-specific binding by blocking the leftover spaces over solid surface after immobilization of a capture biomolecule. However, the acquired diversity of BSA blocking has remained conflicted on nature of the solid surfaces used, antigen–antibody combinations, and their concentrations. Here, we investigate the necessity of BSA blocking in common ELISA protocols by performing ELISA detection of human-IgG, rabbit-IgG, human-IgE, concanavalin A and hepatitis C core antigen with and without BSA blocking on different microplates and with different concentrations of analytes. We found that irrespective of solid surfaces or analyte concentrations, the ELISA protocols with and without-BSA blocking produce similar outcomes when performed with PBST washing. However, if PBS instead of PBST is used for washing in assays with BSA blocking, the chances of wrong predictions enhance significantly. Further, by using FITC-tagged BSA, we have found that BSA binds weakly to microplate surface and escapes during PBST washing. Again, if PBS rather than PBST is used in combination to BSA blocking, case-dependent non-specificity is added to ELISA results. Based on these observations, we suggest to empirically determine the absolute necessity of BSA-blocking, as majority of ELISA protocols do not need BSA-blocking.
Introduction
Enzyme linked immunosorbent assay (ELISA), a mainstream technique in diagnostic molecular science, is preferentially performed in microplates to detect and quantify target analytes.1–3 An ELISA protocol primarily involves the immobilization of antigen followed by its detection using antigen-specific antibody. Antigen immobilization can be performed through direct adsorption (direct-ELISA) or by a capture primary antibody already attached to the surface (sandwich ELISA). In both the procedures, a blocking step is usually included after initial antigen or capture antibody immobilization with an aim to block the residual binding sites on solid surface of the microplates. As ELISA is an important diagnostic tool, non-specific binding of unwanted proteins during the course of assay can give false results. To overcome the problem of non-specific binding, surface of the microplates are frequently blocked with a blocking agent and then washed with a detergent-added washing buffer after each step of a ELISA protocol. The combination of detergents and BSA is itself a controversial issue in ELISA protocol as some reports suggest that the blocking efficacy of Tween-20 enhances in presence of BSA,4 while the commercial suppliers of ELISA kit recommend not to use BSA and Tween-20 together, if used, special care is suggested. Also, evidences exhibit that some specific ELISA protocols, such as the dot blot assay and western blot assay can even be performed in a blocking-free manner by using Tween-20 as an additive to washing buffer.5 Similarly, Li et al., demonstrated an immunoassay for the assessment of cardiac risk by a microfluidic fluorescence heterogeneous point-of-care diagnosis, carried out in blocking-free manner.6 However, as these findings lack comprehensive studies, they failed to capture wide recognition and therefore, a separate blocking step is still practiced worldwide irrespective of its requirement in common ELISA protocols. Among the various protein-based blockers, bovine serum albumin (BSA), fetal bovine serum, skimmed milk casein and ovalbumin proteins are the most commonly used blocking agents.7–11 Owing to the fact that no single blocking reagent is ideal for all ELISAs, selection of an appropriate blocking agent is generally required in a case-dependent manner (antigen–antibody used). This in turn depend on factors such as the nature of antigen–antibody and the solid support used, concentrations of analytes and the incubation times. Except for assay where antibodies are raised by analyte-BSA as immunogen, BSA is the most preferred blocking agent in ELISA applications. However, cases of significant cross reactivity are reported frequently because of the bovine IgG contamination of commercially available BSA.12 Besides, lot-to-lot inconsistency and variability, and reduction in antigen–antibody interactions through steric hindrance or by the endogenous enzyme activity are among the major drawbacks of using BSA in ELISA protocols.13–19 Despite of these prevalent shortcomings, BSA is a well-practiced blocking agent among researchers and clinicians. We reasoned that one of the factors responsible for these ongoing practices could be the lack of perspective studies on efficiency, mechanism and necessity of BSA blocking in commonly used ELISA protocols.Thus, to determine if BSA blocking is an absolute requirement of ELISA, the present study investigates conventional and rapid ELISA protocols for the requisite of BSA blocking. The study is performed on different microplates (polypropylene, polystyrene and polycarbonates) using different combinations and concentrations of antigen–antibodies, analytes and washing conditions. Also, by analyzing the blocking efficiency of BSA on different microplates and under different washing conditions, we demonstrate that BSA binds weakly to the surfaces of microplates and escapes during PBST washings. The findings of present study provide a better description on BSA blocking by comparing the efficiency, necessity and consequences of BSA blocking in common ELISA protocols.
Experimental
Reagents
Polystyrene (Cat. no. 655001), polypropylene (Cat. no. 655207) and polycarbonate (Cat. No. 651585G) medium binding 96-well microtiter plates were purchased from Greiner Bio-One, Germany. Human IgG (Cat. no. I4506), rabbit IgG (Cat. no. I5006), hepatitis C virus core antigen (Cat. no. H9034), concanavalin A (Cat. no. L7647), anti-human IgG (Cat. no. I3382), anti-rabbit IgG (Cat. no. R2004) and anti-concanavalin A (Cat. no. C7401) were purchased from Sigma Aldrich, USA. Human IgE ELISA Quantitation set (Cat. no. E80108) was purchased from Bethyl Laboratories, Inc., US. Phosphate buffer saline (PBS) was prepared by adding 0.85% NaCl to 10 mM phosphate buffer (pH 7.2). PBS-Tween-20 (PBST) was prepared by adding 0.1% Tween-20 (Cat. no. 17131601; Biophamacia Biotech) to PBS. 2% solution of BSA (Cat. no. A7030; Sigma Aldrich) and FITC-BSA (Cat. no. A9771; Sigma Aldrich) in PBS were used as blocking buffers. Horse radish peroxidase (HRP) substrate-dye buffer was prepared by dissolving 6 mg of OPD in 6 mL of citrate buffer, pH 5 and 8 μL of 30% H2O2. Fluorescence measurements were performed in black polypropylene, 96-well microplates (Cat. no. 655209; Greiner Bio-One, Germany). The microplates were activated for the immobilization of coating antibodies using 1-fluoro 2-nitro 4-azidobenzene (FNAB) as reported earlier.20Conventional sandwich ELISA protocol
Conventional sandwich ELISA experiments were performed using standard 18 h protocol. Respective capture antibodies (anti-HIgG, anti-RIgG, anti-HIgE, and anti-lectin; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution from 1 mg mL−1) were coated to PP, PS and PC microplates by overnight incubation at 4 °C. Effect of BSA blocking was examined by performing two distinct set of experiments, with and without BSA blocking. Blocking (2% BSA) was performed for 1 h at 37 °C followed by washing consisting of three consecutive PBST washes to remove excess BSA. Experiments of without-BSA blocking were performed with PBS incubation instead of BSA. Thereafter, the respective analytes (HIgG, RIgG, HIgE and Con A) were added to plates and incubated for 4 h at RT or overnight at 4 °C. The analyte concentration dependent studies were performed with 5 ng mL−1, 50 ng mL−1 and 500 ng mL−1 of each analyte. Unbound or non-specifically bound analytes were removed by 3 washes of PBST, each consisting of ∼5 min incubation in buffer. This followed the binding of respective detection antibodies (1:3000 dilutions; anti-HIgG-conjugate, anti-RIgG-conjugate, anti-lectin conjugate; and anti-HIgE-conjugate) for 1 hour at 37 °C and subsequent PBST washing to remove excess of detection antibody. Colour development was carried out by adding 100 μL of HRP substrate-dye to respective well. After 5 min of incubation at RT, the colour development reaction was stopped by adding 20 μL of 5% H2SO4 to each well. Absorbance of the developed colour was measured at 490 nm. All experiments were performed in triplicates.
:100 dilution from 1 mg mL−1) were coated to PP, PS and PC microplates by overnight incubation at 4 °C. Effect of BSA blocking was examined by performing two distinct set of experiments, with and without BSA blocking. Blocking (2% BSA) was performed for 1 h at 37 °C followed by washing consisting of three consecutive PBST washes to remove excess BSA. Experiments of without-BSA blocking were performed with PBS incubation instead of BSA. Thereafter, the respective analytes (HIgG, RIgG, HIgE and Con A) were added to plates and incubated for 4 h at RT or overnight at 4 °C. The analyte concentration dependent studies were performed with 5 ng mL−1, 50 ng mL−1 and 500 ng mL−1 of each analyte. Unbound or non-specifically bound analytes were removed by 3 washes of PBST, each consisting of ∼5 min incubation in buffer. This followed the binding of respective detection antibodies (1:3000 dilutions; anti-HIgG-conjugate, anti-RIgG-conjugate, anti-lectin conjugate; and anti-HIgE-conjugate) for 1 hour at 37 °C and subsequent PBST washing to remove excess of detection antibody. Colour development was carried out by adding 100 μL of HRP substrate-dye to respective well. After 5 min of incubation at RT, the colour development reaction was stopped by adding 20 μL of 5% H2SO4 to each well. Absorbance of the developed colour was measured at 490 nm. All experiments were performed in triplicates.
Conventional ELISA protocol with varying concentrations (1:100 to 1:10000 dilution) of coating antibody (anti-HIgG, anti-RIgG, anti-lectin and anti-HIgE) were performed in a similar manner on PP, PS and PC 96-well microplates. All the steps of ELISA were performed similar to above mentioned procedure.
Heat-mediated ELISA protocol
Heat-mediated rapid ELISA experiments were carried out by 4 h in a dry heat incubator, as reported earlier.21 Briefly, respective capture antibodies (1:100 dilution from 1 mg mL−1 stock) were coated onto ELISA plates by 1 h incubation at 50 °C. Blocking was carried out with 2% BSA at 45 °C for 1 hour. Experiments without BSA-blocking were performed by adding PBS to antibody coated wells and incubating at similar thermal conditions. Blocked wells were washed thrice by PBST to remove excess BSA. Thereafter, respective analytes (50 ng mL−1; HIgG, RIgG, HIgE, Con A and HCVcAg) were added to the blocked and non-blocked wells and incubated at 50 °C for another 1 hour. Three washes, each comprising ∼5 min incubation in PBST were performed to remove non-specifically adhered analytes. Next, detection antibodies (1:3000 dilution) were added to the respective wells and incubated at 50 °C for 1 h. After washing with PBST, the colour development reaction was carried out by adding 100 μL OPD solution to each well and incubating at RT for 5 minutes. Reaction was stopped by adding 5% H2SO4 and the absorption of the developed colour was measured at 490 nm.
Ultrasound-mediated ELISA protocol
Ultrasound-mediated rapid ELISA experiments were carried out by an already standardised 40 min protocol, in a sonicator water-bath operating at optimum output power of 120 W.22 All the steps of ELISA were performed similar to that of heat-mediated ELISA, except the incubation time which was reduced to 10 min for each step and performed in a sonicator water-bath.Blocking efficiency of BSA and FITC-BSA on PP, PC and PS surfaces
The comparison of BSA to FITC-BSA for their blocking efficiencies were made by performing ELISA detection of RIgG on PP, PS and PC microtiter plates, wherein the blockings were carried out separately with 2% BSA and 2% FITC-BSA. All the steps of assay were performed conventionally using 1:100 dilution of coating antibody, 50 ng mL−1 test analyte and 1:3000 dilution of detection antibody.
Interaction of BSA with solid supports in ELISA protocol
FITC-tagged BSA (2% in PBS) having approx. 7–12 moles of FITC per mole of BSA was added to black PP microtiter plates and incubated for 1 h at 37 °C. After stipulated time interval, the BSA blocked wells were washed separately by PBS and PBST, in two different set of experiments. Washings were performed corresponding to conventional washing carried out after blocking, analyte binding and detection antibody binding (each round of washing consists three washes carried out one after another). Residual fluorescence in the wells after each round of washing was measured at 480–520 nm excitation–emission in fluorimeter (TECAN Infinite 200 PRO, Switzerland). Wells blocked with FITC-BSA with no washing were included as control to monitor the quenching-resulted decrease in fluorescence. The obtained values were normalized and plotted.Interaction of BSA with assay reagents in ELISA protocol
The possible interactions of BSA with assay components such as capture antibody, analytes, or detection antibody was examined by performing ELISA quantification of HIgG, RIgG, HIgE, and Con A in BSA-free and with-BSA conditions using PBS and PBST washes separately. For this, 2% FITC-BSA was used as blocking agent and the assay was performed in black PP microplates. Four set of ELISA experiments: BSA-blocking and PBS washing (BSA + PBS), BSA-blocking and PBST washing (BSA + PBST), no BSA-blocking and PBS washing (only PBS), and no BSA-blocking and PBST washing (only PBST) were carried out to ascertain the effect of BSA blocking and washing on ELISA outcome. All experiments were performed with equivalent amount of primary coating antibodies (1:100 dilution form 1 mg mL−1), analytes (50 ng mL−1) and detection antibodies (1:3000 dilution). After each step of washing, the residual fluorescence of FITC-BSA were measured, and after normalizing with control FITC-BSA, the fluorescence intensities were plotted against individual washing round.
Statistical analysis of the data
Kruskal–Wallis test was performed to determine the difference in ELISA outcome for HIgG, RIgG, HIgE, and Con A when performed with and without BSA blocking. Graphics were drawn using GraphPad Prism (version 5.03 for Windows, GraphPad Software, San Diego California USA).Results and discussion
BSA-blocking step in conventional sandwich ELISA is not essential
Necessity of the BSA-blocking in conventional sandwich ELISA was determined by performing ELISA detection of HIgG, HIgE, RIgG, and Con A at low (5 ng mL−1), medium (50 ng mL−1) and high (500 ng mL−1) concentrations of analytes, with and without-BSA blocking on PP, PS and PC microtiter plates. Washing after different rounds of incubation were performed with PBST buffer containing 0.1% Tween-20. Presence of equivalent absorbance values in all the ELISA experiments, when performed with and without BSA-blocking, and with PBST washing, suggests the minimal role of BSA on assay outcomes. It can also be interpreted that PBST alone can overcome majority of non-specific bindings, making the role of BSA an insignificant. Interestingly, these results showed no variations when performed on different solid surfaces used in ELISA. Further, we analyzed the efficiency of BSA blocking in different analyte concentrations. For this, we chose 5 ng mL−1, 50 ng mL−1 and 500 ng mL−1 as low, medium and high analyte concentrations for each of the tested analytes, based on the average value of upper and lower limit of quantifications determined from standard graph of each analyte (Fig. S1†). We then performed Kruskal–Wallis test to assess the differences in obtained ELISA results of with-blocking and without-blocking experiments by assuming that absorption values of low to high concentration analytes don't follow a normal distribution. The results of Kruskal–Wallis test for HIgG (H = 0.923, 1 d.f., P value = 0.3367; significance level = 0.05), HIgE (H = 0.41, 1 d.f., P value = 0.5218), RIgG (H = 0.641, 1 d.f., P value = 0.4233) and Con A (H = 0.923, 1 d.f., P value = 0.3367) showed insignificant differences when carried out with medium analyte concentrations and with and without BSA blocking (Fig. 1). Similar results were obtained for assays performed on PP and PC surfaces using medium analyte concentration.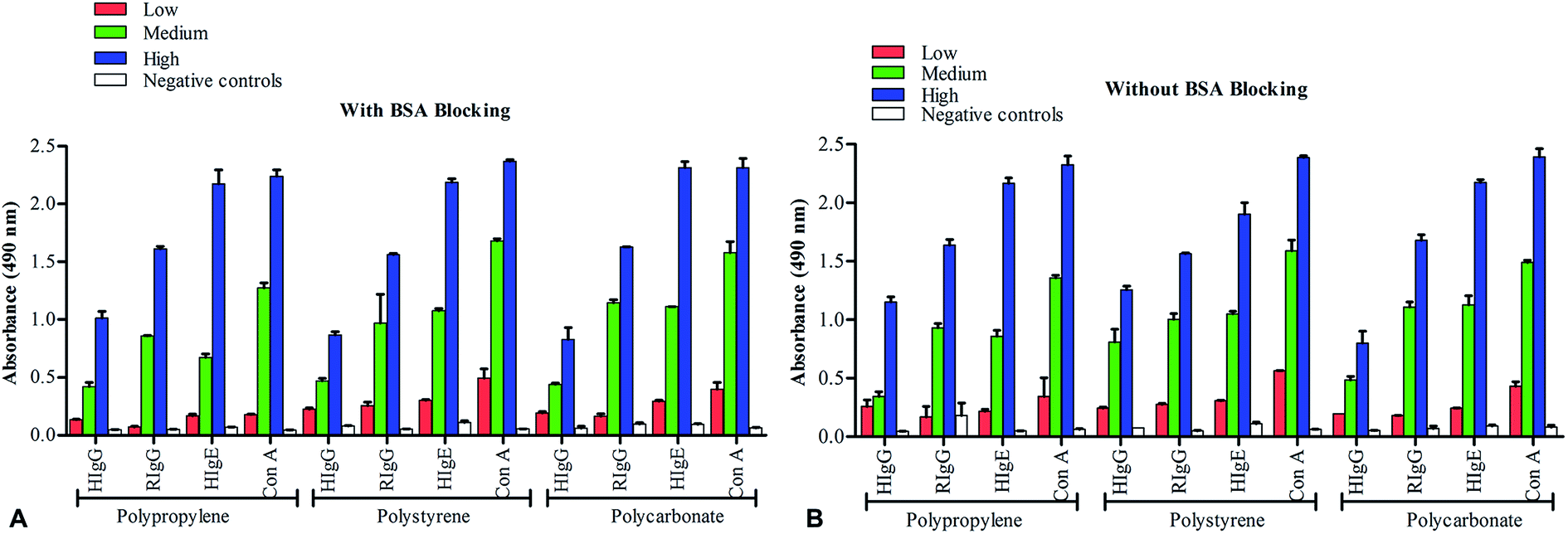 | ||
| Fig. 1 ELISA detection of human IgG, rabbit IgG, human IgE and concanavalin A at different concentrations of analytes, carried out on polypropylene, polystyrene and polycarbonate microplates, with-BSA (A) and without-BSA (B) blockings. Antibody coating (1:100 dilution from 1 mg mL−1), BSA blocking, analyte binding, and detection antibody binding (1:3000 dilution) are performed as per 18 h conventional procedure. Analyte concentrations of 5 ng mL−1, 50 ng mL−1 and 500 ng mL−1 are used as low, medium and high concentrations, respectively. Negative controls (50 ng mL−1; RIgG for HIgG, HIgE and Con A, and HIgG for RIgG) are represented by empty bars. Absorbance values are drawn after subtracting blank. Error bars represent SEM. | ||
In another assessment, we checked if the concentration of primary coating antibody decides the requisite of BSA blocking in ELISA. For this, we used six dilutions ( 1:100 to 1:10000) of primary coating antibodies of each tested analyte and performed ELISA with and without-BSA blockings. As depicted in Fig. 2, decrease in absorbance values with increase in coating antibody dilutions indicates the homogeneity in assay protocol. Besides, the insignificant differences in the results of BSA-free and BSA-blocked ELISA experiments shows the minimal interference by BSA in the ELSIA outcome (Fig. 2). These observations suggest that independent of the analyte concentrations, solid surfaces used or the concentration of primary coating molecules, the BSA-free and BSA-blocking ELISA protocols produces similar results when performed with PBST washes.
 | ||
| Fig. 2 ELISA detection at different concentrations of capture antibody. Detection of HIgG, RIgG, HIgE and Con A on PP, PS and PC surfaces is carried out with-BSA blocking (A) and without-BSA blocking (B) using different dilutions (1:100 to 1:10000 dilution) of coating antibody. | ||
BSA-blocking in rapid ELISA protocols is not required
As observed that BSA plays no significant role in conventional sandwich ELISA protocols; to consider the possible requirement of BSA blocking in rapid ELISA protocols, we have performed 4 h heat-mediated ELISA (HELISA) and 40 min sound-mediated ELISA (SELISA) on PP, PC and PS surfaces, with and without BSA-blocking. We included hepatitis core antigen (HCA) as one more analyte to further increase the diversity of antigen–antibody pairs in our studies. As shown in Fig. 3, the ELISA outcome of HIgG, RIgG, HIgE, Con A and HCA quantification showed no differences when performed with and without BSA blocking steps. Though the obtained absorbance values are slightly low in 40 min SELISA as compared to 4 h HELISA or conventional ELISA, the pattern of absorbance reads in BSA and BSA-free assays have remained constant. This again suggests that independent of assay timings, a BSA blocking step is frequently not required in conventional as well as rapid ELISA protocols when performed with PBST washing buffer.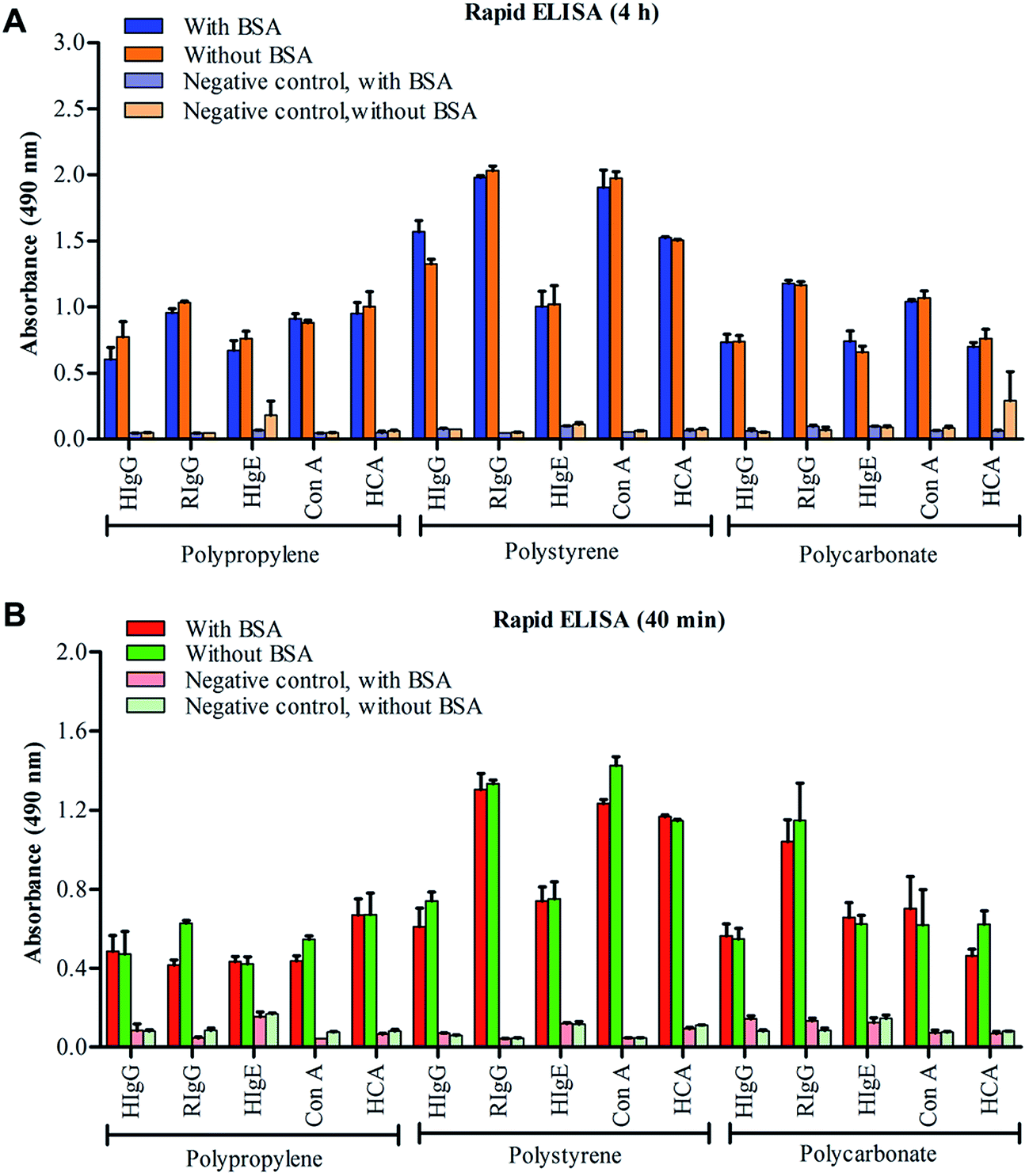 | ||
| Fig. 3 Rapid ELISA protocols. Necessity of BSA blocking in rapid ELISA protocols is determined by performing 4 h heat-mediated ELISA (A) and 40 min sound-mediated ELISA (B) for detection of HIgG, RIgG, HIgE, Con A and HCVcAg on PP, PS and PC microplates. | ||
Probable mechanism of BSA binding: weak interactions of BSA with polymer surfaces result in escape of BSA during PBST washing
In further efforts to determine the efficiency of BSA in blocking the residual binding sites on commonly used microplates in ELISA, we blocked the wells with FITC-tagged BSA and monitored the residual left-over fluorescence after each step of washing in the wells blocked with FITC-BSA. As shown in Fig. 4, the rapid decrease in the fluorescence intensity during the first washing in wells blocked with FITC-BSA suggest the removal of BSA from microplates. Further, this decrease in fluorescence enhance in successive rounds of washing. Interestingly, the loss of fluorescence in PBST washed wells was higher than in the wells washed with PBS buffer. We assumed that the initial decrease in relative fluorescence upon first wash either with PBS or PBST may have resulted from the removal of excess BSA* which might have adhered weakly to the microplate surface. The subsequent loss in fluorescence intensity, which was high in case of PBST washed wells, indeed signify the weak binding and rapid removal of blocking BSA* from solid supports in presence of detergent-added washes. These observations also suggests that the combination of BSA-blocking and PBST washing is not beneficial as it causes rapid loss of blocking BSA from the polymer surface.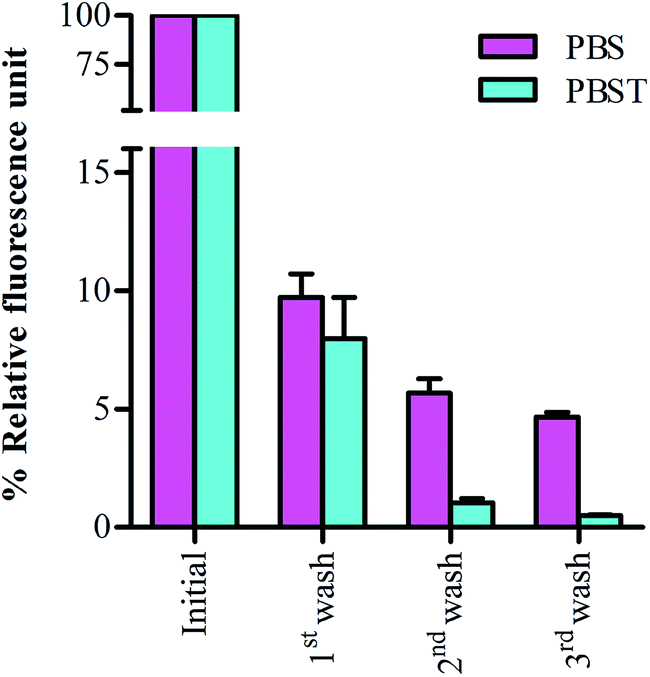 | ||
| Fig. 4 Monitoring the retention of FITC-tagged BSA in microplates. Blocking efficiency of FIT-BSA in presence of PBS and PBST wash is determined by measuring the residual fluorescence of the wells. Washing are performed corresponding to conventional washing carried out after blocking, analyte binding and detection antibody binding. | ||
BSA may interact with assay components in sandwich ELISA experiment and enhances non-specific bindings
The possible interactions among BSA and other assay components were examined by performing ELISA wherein FITC-BSA is used for blocking and the residual fluorescence after each round of PBS/PBST washing were measured. As shown in Fig. 5a, the loss in fluorescence intensity after PBS/PBST washes has followed similar pattern as that of the assay where the wells were blocked with FITC-BSA without any added antigen–antibodies (Fig. 4).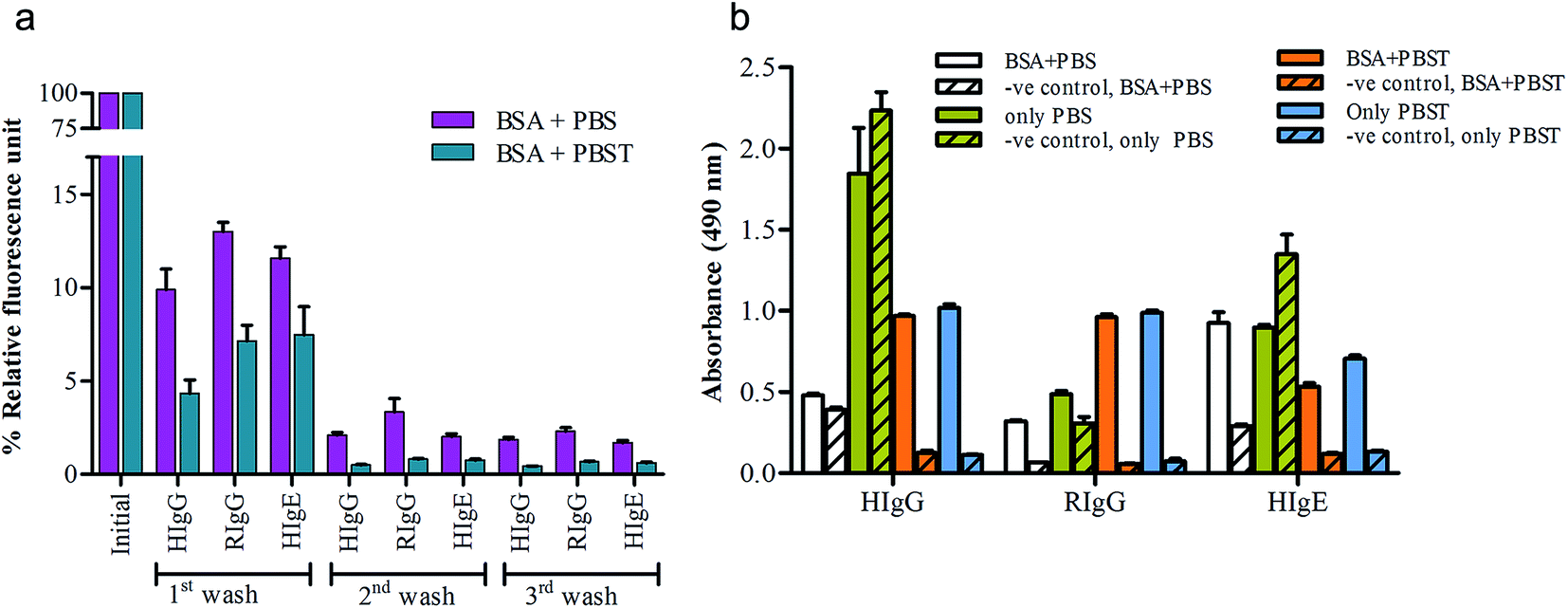 | ||
| Fig. 5 Measuring the cross-talk of BSA with assay components in ELISA. Residual fluorescence in FITC-BSA blocked wells is measured after each round of washing (a). Colorimetric detection of BSA-interactions with assay components is carried out using four set of experiments- (i) BSA + PBS, (ii) only PBS, (iii) BSA + PBST, (iv) only PBST (b). The RIgG is used as negative control for HIgG and HIgE. HIgG is used as negative control for RIgG. | ||
Further, as the retention and escape of BSA from the wells of microplate is found to depend on washing conditions, we performed four set of ELISA experiments for each analyte (HIgG, RIgG, HIgE and Con A) to find out the best procedure that produces optimal results without any non-specificity. The chosen conditions for ELISA experiments were- (i) BSA-blocking and PBS washing, (ii) BSA blocking and PBST washing, (iii) no-BSA blocking and PBS washing, and (iv) no-BSA blocking and PBST washing. As shown in Fig. 5b, the BSA* blocked wells having captured anti-human IgG (for HIgG detection) showed low ELISA values in wells washed with PBS over PBST. Also, the negative control (RIgG in place of HIgG) of the assay showed remarkably high absorbance values. To ascertain if BSA blocking or washing conditions are responsible for such abundant non-specificity, two control experiments were performed where the washings were carried out with PBS and PBST without any BSA blocking (only PBS and only PBST). As depicted in Fig. 5b, compared to previous experiment (BSA + PBS), we obtained high ELISA value in wells washed with PBS without any blocking step. Interestingly, the ELISA values of only PBST experiment were comparable to the BSA + PBST experiment. This signifies that in absence of PBST washing, irrespective of BSA blocking, the chances of getting false results enhances significantly. Yet, if BSA is included (without PBST wash; BSA + PBS), it may resist cross-reactivity, but as same time, can add or enhance other non-specific interactions. However, if same assays are performed with PBST washes, irrespective of BSA blocking (i.e. BSA + PBST or only PBST), the results remain consistent across variety of antigen–antibody combinations.
In further assays for RIgG and HIgE detection, the PBST washing has produced similar outcomes with or without blockings. Based on these observations, we recommend the use of PBST for ELISA washes and not PBS alone. At the same time it is also suggested to empirically determine the necessity of BSA blocking in a case-dependent manner because most of the assays do not need BSA blocking and simple PBST wash (0.1%) can overcome all non-specific interactions.
Conclusion
Blocking in common ELISA assays has remained a controversial topic as some reports suggest that ELISA can be performed in a blocking-free manner, while others believe in an absolute requirement of blocking. In this long quest of divergent beliefs, we have selectively chosen BSA due to its predominance in common ELISA protocols. We have taken in account the variety of factors that may influence the efficiency or requirement of BSA blocking and checked if BSA plays significant role in common sandwich ELISA protocols. Our findings show that the requisite of BSA in ELISA assay depends mainly on the antigen–antibody; however, BSA is not required when the ELISA is performed with the detergent-added washes. The novelty of our finding is the perspective study that we conducted on different combinations of antigen–antibodies, at varying concentrations of capturing antibody and analyte molecules, on different solid surfaces using different washing conditions and different times of incubation. In antigen–antibody combinations, we covered human immunoglobulins, rabbit immunoglobulins, plant allergen and viral proteins, while in solid surfaces, we analyzed the most commonly used polypropylene, polystyrene and polycarbonate microtiter plates. Similarly, three different protocols of ELISA are performed- 18 h conventional ELISA, 4 h rapid HELISA and 40 min SELISA. Further, to decipher the fate and efficiency of BSA-blocking on common microplates surfaces, we have monitored the retention of FITC-BSA and found that the BSA–polymer surface interactions are weak, which result in loss of BSA during PBST washing. At the same time, we also analyzed the efficacy of detergent (Tween-20) in washing buffer to remove non-specifically adhered components or even the loosely bound BSA. We found that inclusion of Tween-20 to washing buffer removes majority of non-specific binding including the transient binding of BSA to the polystyrene surface. If the detergent is not added to washing buffer, the blocking BSA itself may act as a hindering molecule and may produce false results. In conclusion, we suggest that ELISA assays can be performed in blocking-free manner, as the added detergent in washing buffer is effective enough to remove majority of the non-specific bindings. Also, the removal of BSA-blocking step can prove to be an added advantage of reduced ELISA timing and assay-cost.Acknowledgements
The authors declare no competing interests. R. A. thanks Council of Scientific and Industrial Research, Government of India for the award of a senior research fellowship. This work is funded by CARDIOMED project no. BSC0122 of Council of Scientific and Industrial Research, Government of India, New Delhi, India.Notes and references
- B. E. Batteiger, Blocking of Immunoblots, Handbook of Immunoblotting of Proteins, ed. O. J. Bjerrum and N. H. H. Heegaard, CRC Press Inc., Boca Raton, Florida, 1988, vol. I, pp. 145–150 Search PubMed.
- E. Engvall and P. Perlmann, J. Immunol., 1972, 109, 129–135 CAS.
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871–874 CrossRef CAS PubMed.
- P. V. Hornbeck, Enzyme-Linked Immunosorbent Assays, Current Protocols in Immunology, 2015, 110, 2.1.1–2.1.23, DOI:10.1002/0471142735.im0201s110.
- K. Mohammad and A. Esen, J. Immunol. Methods, 1989, 117, 141–145 CrossRef CAS PubMed.
- P. Li, A. J. Sherry, J. A. Cortes, C. Anagnostopoulos and M. Faghri, Biomed. Microdevices, 2011, 13(3), 475–483 CrossRef CAS PubMed.
- D. A. Johnson, J. W. Gautch, J. R. Sportsman and J. Elder, Gene Anal. Tech., 1984, 1, 3–8 CrossRef CAS.
- H. Towbin, T. Staehelin and J. Gordon, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 4350–4354 CrossRef CAS.
- R. V. Vogt Jr, D. L. Phillips, L. O. Henderson, W. Whitfield and F. W. Spierto, J. Immunol. Methods, 1987, 101, 43–50 CrossRef CAS.
- E. S. Sawyer and P. J. Sawyer, Fish serum as a blocking agent, U. S. Patent US005602041A, 1997.
- R. Lange, H. Bocklage, T. Schneider, H. W. Kolmel, J. Heesemann and H. Karch, J. Clin. Microbiol., 1992, 30(1), 229–232 CAS.
- H. Miura, M. Kitano, A. Yoneyama, A. Kuwahara, K. Moriyama and S. Kitajima, Rinsho Byori, 2005, 53(12), 1103–1108 CAS.
- K. L. Ambroz, Y. Zhang, A. Schutz-Geschwender and D. M. Olive, Proteomics, 2008, 8, 2379–2383 CrossRef CAS PubMed.
- Y. Xiao and S. N. Isaacs, J. Immunol. Methods, 2012, 384(1–2), 148–151 CrossRef CAS PubMed.
- N. DenHollander and D. Befus, J. Immunol. Methods, 1989, 122(1), 129–135 CrossRef CAS PubMed.
- W. Y. Graig, S. E. Poulin, M. F. Collins, T. B. Ledue and R. F. Ritchie, J. Immunol. Methods, 1993, 158, 67–76 CrossRef.
- W. Y. Graig, S. E. Poulin, C. P. Nelson and R. F. Ritchie, Clin. Chem., 1994, 40, 882–888 Search PubMed.
- M. Harvey, R. Kremer and L. Vickers, Guide to diagnostic rapid test device components, Shleicher & Schuell, Keene, N.H, 2nd edn, 2000 Search PubMed.
- R. F. Vogt, D. L. Phillips, L. O. Henderson, W. Whitfield and F. W. Spierto, J. Immunol. Methods, 1987, 101, 43–50 CrossRef CAS PubMed.
- U. Bora, L. Chugh and P. Nahar, J. Immunol. Methods, 2002, 268, 171–177 CrossRef CAS PubMed.
- U. Bora, K. Kannan and P. Nahar, J. Immunol. Methods, 2004, 293(1–2), 43–50 CrossRef CAS PubMed.
- P. Sharma and P. Nahar, Anal. Chim. Acta, 2009, 650, 241–246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20750a |
| This journal is © The Royal Society of Chemistry 2015 |