
Structure and thermal properties of phosphorus-containing polyol synthesized from cardanol†
Abstract
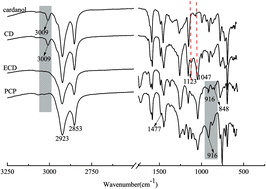
P-Containing cardanol polyol (PCP), a novel cardanol derivative, was synthesized. Its structure was confirmed by Fourier transform infrared spectrometry (FT-IR) and 1H nuclear magnetic resonance (1H NMR). Its flame retardancy and thermal stability were assessed by limiting oxygen index (LOI) and thermogravimetric analysis (TGA). TGA showed that the degradation occurred in two stages with varying mass rate losses of PCP. The PCP showed higher initial decomposition temperature (Ti), final degradation temperature (Tf), and maximum degradation temperature (Tmax) than cardanol diol (CD) of the same class, and also had higher mass residual than CD (6.77% vs. 0.02%). To study the flame retardancy of PCP, we prepared a series of polyurethane foams (PUFs) with different PCP content. LOI increased continuously with the increasing content of PCP added into PUF. The thermal degradation of PCP was disclosed by TGA-FTIR and TGA-MS. During degradation, PCP continued to release PO free radicals and o-phenylphenoxyl free radicals, which acted as scavengers of H˙ and OH˙ during two degradation stages at 200 °C. Moreover, the phosphaphenanthrene group may create a char residue acting as a barrier for the polymer matrix.
 Please wait while we load your content...
Please wait while we load your content...

