
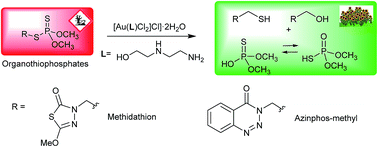
An Au(iii)–amino alcohol complex for degradation of organophosphorus pesticides†
Abstract
An Au(III)–amino alcohol complex has been used to cleave organophosphorous pesticides of the dithiophosphate family. P–S bond breaking was readily demonstrated by 1H NMR, 31P NMR and MS. Thiol fragment release was also demonstrated using 2,4-dinitrobenzenesulfonyl fluorescein ethyl ester as a fluorescent sensor.
 Please wait while we load your content...
Please wait while we load your content...

