
Comprehensive investigation of the reciprocity of structure and enhanced photocatalytic performance in finned-tube structured TiO2/BiOBr heterojunctions†
Chao Xuea,
Xin Xub,
Guidong Yang*a and
Shujiang Ding*b
aDepartment of Chemical Engineering, School of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an, 710049, P. R. China. E-mail: guidongyang@mail.xjtu.edu.cn
bDepartment of Applied Chemistry, School of Science, State Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an, 710049, P. R. China. E-mail: dingsj@mail.xjtu.edu.cn
First published on 17th November 2015
Abstract
Finned-tube structured TiO2@BiOBr heterojunctions were synthesized via a facile and efficient solvothermal process. Field-emission scanning electron microscopy and transmission electron microscopy analyses demonstrated that the tunable uniform lamellar structured BiOBr nanoplates burgeoned from the surface of one-dimensional TiO2 nanotubes and constructed intimate interfacial junctions. The formation mechanism of the as-prepared TiO2@BiOBr heterojunctions was thus proposed. Owing to the combined effects of the intense visible light absorption, the efficient charge separation and lower recombination of photo-generated electron–hole pairs as well as larger specific surface area, the as-fabricated heterojunctions exhibited the best visible-light photocatalytic activity, structural stability and sustained cycling performance compared with the reported congeneric catalysts. Their degradation rate remained at 95.5% after seven cycles for the photodecomposition of RhB. A possible mechanism of the photocatalytic activity enhancement was also proposed based on the photocurrent measurements, the photoluminescence analysis and the radical trapping experiments. It revealed that the intimate interfacial junction could promote the separation of photo-generated charge carriers, simultaneously, both ·O2− and h+ acted as the main reactive species in the rapid degradation of RhB under visible-light irradiation.
1 Introduction
Over the past few decades, titanium dioxide (TiO2) has attracted considerable attention because of its widespread application in pollution abatement and exploitation of new energy resources. However, some of its intrinsic drawbacks greatly limit its application. For instance, the sunlight utilization efficiency of TiO2 is extremely low due to the restriction of a large bandgap (Eg = 3.2 eV for anatase).1,2 Additionally, the high recombination rate of the photo-generated electron–hole pairs in the TiO2 also leads to lower quantum efficiency. To solve the problem above, many strategies are mainly focusing on the design and synthesis of various novel composite photocatalysts through incorporating TiO2 with high performance visible-light-responsive semiconductors. Recently, owing to the unique layered structure, outstanding optical and electrical properties as well as narrower band gap (about 2.9 eV), BiOBr is considered to be one of the best ideal components for tailoring TiO2 nanostructures and extending the absorption of TiO2 into the visible light range.3–8 And thus an increasingly great number of BiOBr hybrids have been synthesized for various applications. For example, Cao's group designed a family of bifunctional sunlight-driven photocatalysts and the BiOBr@TiO2 framework exhibited outstanding sunlight-driven photocatalytic activity.9 Wu's group prepared a flake-tube structured BiOBr–TiO2 nanotube array heterojunction via the S-CBD method and the as-prepared sample exhibited remarkably enhanced photocatalytic activity.2 Wei et al. fabricated a hybrid BiOBr–TiO2 nanocomposite with high surface area and mesoporous structure by one-pot solvothermal approach. The resultant catalyst showed a distinguished photocatalytic performance.10Nevertheless, although a great deal of efforts have been devoted to improve the photocatalytic activity of BiOBr/TiO2 composites, the photodegradation ability and the practical applications of BiOBr hybrids are not satisfying. For one thing, BiOBr could only absorb partial visible light due to its relatively wide band gap.6,7,11 For another, most BiOBr/TiO2 composites have been created by simply mixing with each other and only a diffuse interface was formed between the two semiconductor candidates in the mixing process, as a result, the charge transfer in the composite would be not spatially smooth as expected. Furthermore, the suspended granular catalysts are easily lost and it may cause secondary pollution which greatly restricted its practical application.12,13 To date, however, it is still extremely challenging to find an efficient synthetic method for fabricating TiO2@BiOBr heterojunction composites with a closely contacted interface and highly photocatalytic activity. Therefore, a promising approach to overcome the current drawbacks of TiO2@BiOBr hybrids is constructing new nanostructured composites with well-defined morphologies, such as one-dimensionally (1D) tubular structured photocatalyst,14 which has been widely used as an ideal nanostructure in photocatalytic applications. This kind of material not only show distinct chemical and unique structural behavior and ascendant photocatalytic activity compared with those of bulk materials,15 but also can provide a large specific surface area as well as beneficial for capturing the light and facilitating the mass transfer of contamination. Moreover, constructing a heterojunction between 1D TiO2 and tiny BiOBr nanoplate will accelerate the separation of photo-generated electron–hole pairs through the contacted interface in the junction and diffuse the catalytical active sites at the semiconductor/liquid interface where they drive the photoreaction.1,2 To the best of our knowledge, rare literatures have focused on 1D nanotubes consisting of TiO2 and BiOBr coupling systems.
Herein, we report a new facile and efficient method for preparing a finned-tube structured TiO2@BiOBr heterojunction (TBNTs) where the tunable uniform lamellar structured BiOBr nanoplates burgeoning from the surface of 1D TiO2 nanotubes (NTs). The strategy consists of a simple cationic polymerization for fabrication crosslinked polymer nanotubes which used as hard template for preparation of 1D TiO2 NTs and subsequent solvothermal synthesis of TBNTs. Importantly, the morphology and size of two-dimensional (2D) BiOBr nanoplates can be well tuned by varying the reaction time and additional surfactant during the process of solvothermal. Notably, the as-prepared TBNTs sample exhibits exceptional visible light photocatalytic activity and cycling capacity than the mechanical mixture of TiO2–BiOBr, pure BiOBr nanoplates and TiO2 NTs.
2 Experimental section
1D TiO2 NTs were fabricated via sol–gel method and a subsequent annealing process. And the 1D finned-tube structured TiO2@BiOBr heterojunctions (TBNTs) were constructed by a new facile and efficient solvothermal method. The fabrication procedure, characterization, general materials and instruments used in this work are described in more detail in the ESI.†3 Results and discussion
Fig. 1a schematically illustrates the formation mechanism of the 1D finned-tube structured TiO2@BiOBr heterojunctions. Firstly, the crosslinked bamboo-like polydivinylbenzene nanotubes (PNTs) were fabricated directly by a fast ultrasonic assisted spontaneous cationic polymerization at room temperature.16,17 It can be seen from the scanning electron microscopy (SEM) that the as-prepared PNTs are several micrometres long and entangle into a network. In addition, the PNTs possess quite smooth surface and have a mean exterior diameter 100–150 nm (Fig. 2a). The transmission electron microscopy (TEM) image further indicates that the PNTs are segmented by the periodic nodes which formed a bamboo-like tubular structure and the surface of the PNTs is extremely smooth (Fig. 2b). Noted that the inter node of PNTs can offer enhanced mechanical strength which is beneficial to the PNTs serving as hard template for the fabrication of 1D TiO2 NTs. Afterwards, the uniform sulfonated PNTs (SPNTs) have been successfully obtained by sulfurization which introducing the sulfonic acid groups (–HSO3) onto the surface of PNTs (Stage I, Fig. 1a). This process is a crucial advantage for synthesizing the TiO2/SPNTs coaxial nanocables,18,19 and the positively charged titanium precursor ions can be easily adsorbed on the surface of the SPNTs due to the electrostatic interaction with the negative functional groups (–HSO3).13 Benefited from this, gradual attachment of a layer of titanate precursor made the surface of SPNTs become relatively rougher than that of PNTs (Fig. 2c). The TEM image clearly reveals that the obtained TiO2@SPNT composite with an increased average diameter of 120–175 nm after cladding a layer of TiO2 precursor and the typical thickness of the TiO2 layer is approximately 15–25 nm (Fig. 2d). Subsequently, a calcinations procedure was conducted to remove the PNT templates and acquired well crystalline tubular TiO2 nanostructure (Stage II, Fig. 1a). Then the TBNTs can be obtained after a solvothermal process (Stage III, Fig. 1a), however, in order to facilitate the growth of BiOBr nanoplates on surface of TiO2 NTs, some indispensable measures were adopted to control the size of BiOBr nanoplates and prevent its self-assemble into microspheres. Fig. 1b reveals the growth behaviour of the BiOBr nanoplates in detail. As shown, the TiO2 NTs were surface modified by a certain concentration of sulfuric acid and subsequently calcined to form the substantial double chelating ligand structure. As a result, the existence of special chelating ligand structure makes the surface of TiO2 NTs show negative charge, which leads to a comfortable nucleation and growth environment for BiOBr nanoplates. Afterwards, the desired 2D crystalline BiOBr nanoplates were controllably obtained in a large-scale by simply solvothermal treatment followed by adjusting the reaction time. Noted that the ethylene glycol (hereafter EG) solution was selected as the solvent which can really help to prevent the premature reaction of Bi3+ cations with the halogen ions.9 This is because the EG could coordinate with Bi3+ cations to generate alkoxide complexes (Bi(OCH2CH2OH)2+) (hereafter Bi(OR)2+). During the solvothermal, the strong coulomb coupling force accelerates the positively charged Bi(OR)2+ ions toward to the negatively charged chelating ligand structure and tend to nucleate and grow on the surface of modified TiO2 NTs. During nucleation, the linearly aligned structured Bi(OR)2+ served as stable nucleation sites which can long-lasting supply of Bi3+ cations with a releasing rate and thus slow down the growth rate of BiOBr crystals.8 During the growth process, cetyltrimethyl ammonium bromide (hereafter CTAB) was added into the solution, which not only served as surfactant, but also was a Br-containing compound to provide Br source. The combination occurred between Br− and Bi(OR)2+ led to the oriented attachment of 2D BiOBr nanoplates on the surface of modified TiO2 NTs, resulting in the fabrication of the finned-tube structured TiO2@BiOBr heterojunctions.20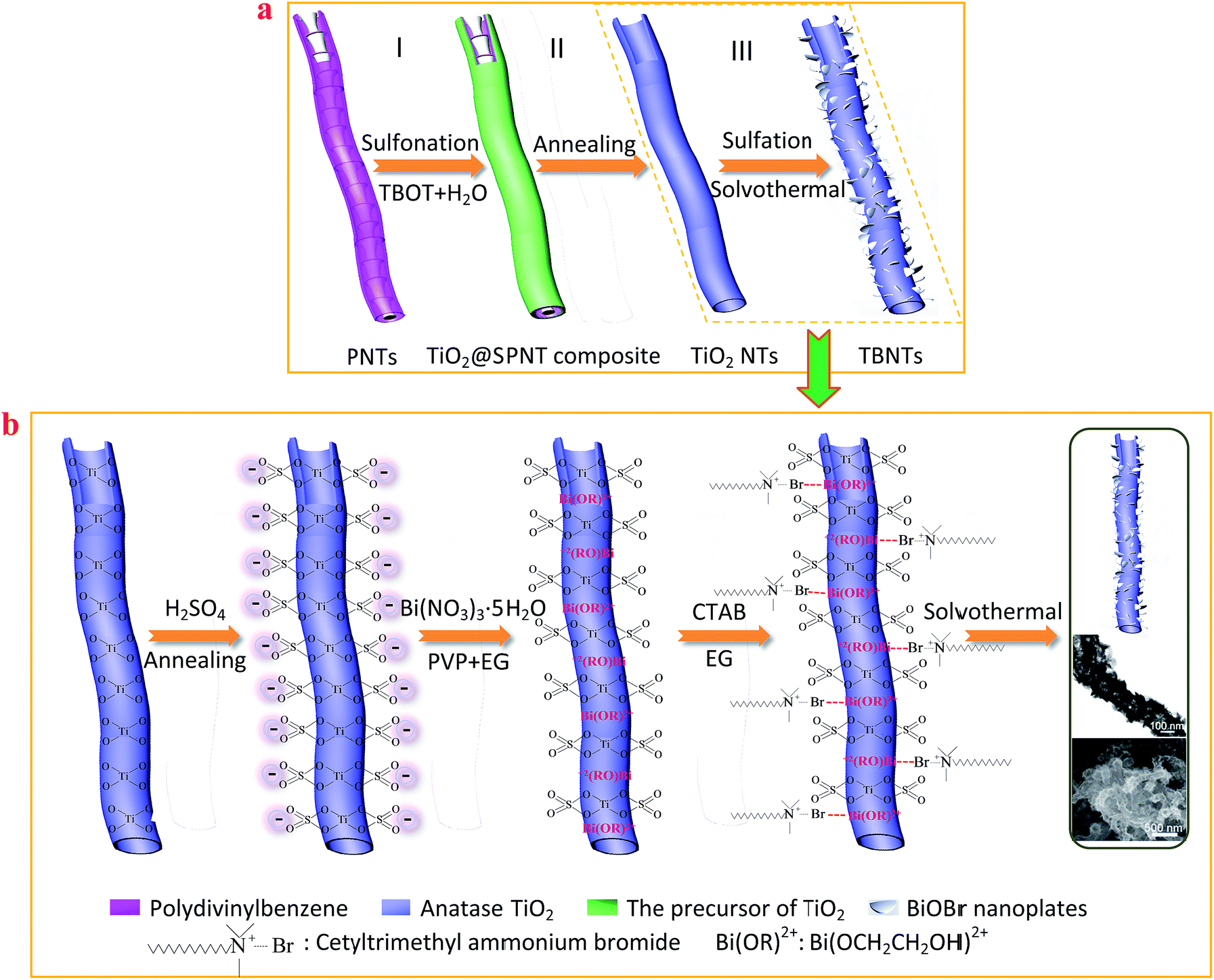 | ||
| Fig. 1 (a) Schematic diagram illustrating the synthetic route of one-dimensional finned-tube structured TiO2@BiOBr heterojunction. (b) The growth behaviour of the BiOBr nanoplates. | ||
 | ||
| Fig. 2 (a) SEM image and (b) TEM image of PNTs; (c) SEM image and (d) TEM image of TiO2@SPNT composite. | ||
Also noteworthy is that the growth behaviour of the BiOBr nanoplates were significantly affected by poly(vinylpyrrolidone) (hereafter PVP) and the solvothermal time, simultaneously. The presence of PVP surfactant resulted in the smaller size of BiOBr nanoplates. On the one hand, the long polymeric chain structured PVP could be served as the capping agent in the reaction system. It could completely surround one or more tiny crystalline nuclei of [Br–Bi–O–Bi–Br] slices resulting in the oriented growth and assembly of nucleation by nonbonding van der Waals force through the halogen atoms along c-axis.21 On the other hand, the surface stabilizer PVP would selectively adsorb onto the surfaces of BiOBr and significantly inhibit the random aggregation of individual nanoplates owing to the stereo-hindrance effect. Noted that, with the prolonged ripening time, the individual nanoplates will gradually assemble together via Ostwald ripening and ultimately constructed three-dimensional (3D) BiOBr microspheres configurations (Fig. S1c–f†).8,20–23
The morphology and microstructure of the resultant TBNTs were detected by SEM, TEM and high-resolution transmission electron microscopy (HR-TEM). As demonstrated in Fig. 3a–c, a large amount of lamellar structured BiOBr nanoplates have uniformly burgeoned from the outer walls of modified 1D TiO2 NTs and successfully constructed the unique finned-tube structured TiO2@BiOBr heterojunctions (TBNTs-1) after solvothermal reaction for 30 min. It is obvious that most of these tiny nanoplates were perpendicular to the TiO2 NTs with only 10 nm in thickness and about 70–120 nm in plane size. The images of TEM further confirm that the TBNTs-1 sample maintain well-defined morphologies after relatively regular anisotropic growth of substantial nanoplates (Fig. 3d–f). It can be seen from Fig. 3d and e that the obtained TBNTs-1 possessed the smooth inner channels and open ends. The close contact between BiOBr nanoplates and TiO2 NTs indicates that a compact junction was constructed in the TiO2@BiOBr composites (Fig. 3f). The corresponding selected area electron diffraction (SAED) pattern (inset of Fig. 3e) shows that the TBNTs-1 sample has the polycrystalline nature and owns well-defined crystallinity.12 The HRTEM image (Fig. 3g) clearly reveals a very close interface between BiOBr nanoplates and TiO2 NTs in tightly coupled system, which further confirms the novel composites photocatalyst possesses the intimately interfacial junction rather than a physical mixture of two separate phases of BiOBr and TiO2. Furthermore, the interplanar spacing of 0.354 nm and 0.405 nm could be indexed to (101) and (002) plane of tetragonal phase of BiOBr crystal (JCPDS no. 09-0393), respectively. Meanwhile, the lattice space of 0.352 nm is in good agreement with (101) crystal plane of anatase TiO2 (JCPDS no. 21-1272). Notably, the TBNTs-1 consisting of the hollow nanotubes and nanoplates not only provides a large specific surface area, but also reduces the scattering of incident light as well as facilitates the mass transfer of contamination.24 Moreover, the intimately interfacial junction facilitate photo-induced charge transfer and restrain photo-generated electron–hole pairs recombination.8,22,23,25 Also, the finned-tube structure is beneficial for the capture of visible light photons and improves the light response ability. Thus, the synthesized sample can exhibit high photocatalytic performance under visible light irradiation.
 | ||
| Fig. 3 (a–c) SEM images; (d–f) TEM images and (g) HR-TEM images of the TBNTs-1 sample. | ||
In order to explore the optimum reaction conditions, a series of time-dependent experiments were performed, and the resulting samples prepared at 160 °C for 45 min, 60 min and 120 min are denoted as TBNTs-2, TBNTs-3 and TBNTs-4, respectively (Fig. S1†). Additionally, the sample fabricated by solvothermal treatment at 160 °C for 30 min, but without adding PVP surfactant, was named as TBNTs-5 (Fig. S2†). For comparison, the pure 2D BiOBr nanoplates aggregates and the mechanical mixture of TiO2–BiOBr (hereafter MTB) were also prepared (see ESI†). As shown in Fig. S1a and b,† when the reaction time is lengthened to 45 min, the crystal size of the BiOBr nanoplates gradually grown and intensively coated on the surface of nanotubes with an average thickness about 30 nm which was much larger than that of TBNTs-1. Moreover, as the ripening time prolonged, both of the TBNTs-3 (Fig. S1c and d†) and TBNTs-4 (Fig. S1e and f†) samples show the similar morphology profiles, which were composed of numerous well-defined 3D flower-like BiOBr microspheres with different diameters and many individual 1D TiO2 NTs. Simultaneously, the 3D flower-like BiOBr microspheres were constructed with irregular assembled BiOBr nanoplates which occupied significantly larger size comparing with the first two samples TBNTs-1 and TBNTs-2. It is believed that under the effect of Ostwald ripening, the BiOBr nanoplates stacked one over another and finally fabricated the microsphere architectures. As obviously presents in Fig. S2,† 1D TiO2 NTs were radically wrapped and tightly adhered onto the surface of 2D BiOBr nanoplates forming the TiO2@BiOBr hybrids (TBNTs-5 sample). It can be attributed to that the absence of PVP would lead to the random growth and gradual aggregation of the crystalline nuclei, subsequently make the [Br–Bi–O–Bi–Br] slices stack together and generated the individual nanoplates during solvothermal process. Moreover, it can be observed that the morphology of BiOBr nanoplates in TBNTs-5 sample were similar to that of pure 2D BiOBr nanoplates aggregates (Fig. S3a and b†), which have the varying size ranging from 300 to 500 nm and approximately 10–20 nm in thickness. In short, the growth process of the finned-tube structured TiO2@BiOBr heterojunctions can be divided into the following four steps: (1) diffusion and absorption (2) nucleation; (3) anisotropic growth and (4) Ostwald ripening. According to the explanation of the growth mechanism, the reaction time and solvent composition should be critical contributors which can be easily adjusted to control the configuration of the products.
The XRD patterns in Fig. 4 show that the samples are composed of anatase TiO2 (JCPDS no. 21-1272) and tetragonal BiOBr (JCPDS no. 09-0393).26,27 It can be observed that the relative diffraction intensities of the BiOBr XRD peaks in the TBNTs-1 sample are distinctly much stronger than that of MTB sample, indicating that well crystallized TiO2@BiOBr heterojunctions can be fabricated by simple strategy of solvothermal. It was also surprising to note that a new diffraction peak at 28.8° appeared both in the TBNTs-1, MTB and pure BiOBr samples which was indexed to (304) crystal plane of single phase Bi24O31Br10 sample (JCPDS no. 03-0564). The existence of small amounts of Bi24O31Br10 may help to improve the photocatalytic activity of the sample considerably, due to its narrow band gap (about 1.7 eV).28 Furthermore, the analysis of Energy Dispersive X-ray Spectrum (EDX, Fig. S4†) reveals that the atomic ratio of Br/Bi in TBNTs-1 sample was approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5.29 As for MTB sample, the atomic ratio of Br/Bi was nearly 7:1 resulting from insufficiency of mechanical mixing. The similarities of these two samples are that a large amount of O element was also detected in EDX spectrum due to the co-existence of TiO2 and BiOBr. In additional, the possible functional groups of the resultant samples were also characterized by Fourier Transform Infrared (FT-IR) Spectroscopy (Fig. S5†). The result of FT-IR spectra analysis is consistent with that of the EDX analysis.30–38
:1.5.29 As for MTB sample, the atomic ratio of Br/Bi was nearly 7:1 resulting from insufficiency of mechanical mixing. The similarities of these two samples are that a large amount of O element was also detected in EDX spectrum due to the co-existence of TiO2 and BiOBr. In additional, the possible functional groups of the resultant samples were also characterized by Fourier Transform Infrared (FT-IR) Spectroscopy (Fig. S5†). The result of FT-IR spectra analysis is consistent with that of the EDX analysis.30–38
 | ||
| Fig. 4 XRD patterns of all samples. | ||
The finned tubular structure and the chemical composition of TBNTs-1 sample were further confirmed by scanning transmission electron microscopy (STEM) and EDX elemental mapping spectroscopy. As intuitively displayed in Fig. 5a and b, the as-formed TiO2@BiOBr composites are composed of the internal tubular structure and uniform layer of the approximate triangular cross section nanoplates which tightly loaded on the outer surface of the nanotubes. Furthermore, EDX elemental mapping images unambiguously demonstrated the homogeneous spatial distribution of Ti (red), O (orange), Br (green) and Bi (bluish violet). As shown in Fig. 5c, Ti was solely located at the concentrated distribution area with smooth edges (Fig. 5c). In contrast, Br and Bi elements had the analogous distribution with the large diameter distribution along the coaxial direction and relatively rough edges (Fig. 5e and f). This can be interpreted by the facts that Ti element came from TiO2 NTs, while Br and Bi elements primarily stemmed from BiOBr nanoplates which wrapped on the surface layer of 1D TiO2 NTs. As for oxygen element, it can be derived from the two substances mentioned above (Fig. 5d). Interestingly, the same phenomenon can be found in the segment of cracked tubular structure, simultaneously. Besides, the associated EDX spectrum analysis (Fig. 5g) further confirmed that the formation of TiO2@BiOBr heterojunctions with Ti, O, Br and Bi as the primary elemental components. Furthermore, the atomic ratio of Br and Bi is about 1:1.2. Thus, it provides confident evidence to confirm that the finned-tube structured TiO2@BiOBr heterojunction was successfully fabricated.
 | ||
| Fig. 5 STEM-EDX elemental mapping and of EDX spectrum TBNTs-1 sample. (a) TEM image. The corresponding EDX elemental mapping images of (b) TBNTs-1 sample (full color); (c) titanium (red); (d) oxygen (orange); (e) bromine (green) and (f) bismuth (bluish violet) in the selected area; (g) EDX spectrum, respectively. | ||
As shown in Fig. 6A, all the samples exhibit quite different absorptions in the UV-visible light region. It is clear to see that the sulfated TiO2 NTs and P25 TiO2 have almost no absorbance in the visible light range with an absorbance edge at 390.2 nm and 398.4 nm, respectively, whereas pure BiOBr nanoplates exhibited a strong light response in both ranges of ultraviolet and visible light with obvious absorption edge at about 460.3 nm. In comparison with the modified TiO2 NTs and P25 TiO2, the UV-Vis absorption edges of TBNTs-1 and MTB samples have obvious red shift which tempestuously extended to about 456.0 nm and 447.7 nm, respectively, indicating its possible application as visible-light-driven photocatalyst. The cause of this phenomenon can be ascribed to the presence of BiOBr nanoplates which has a relatively intrinsic narrow energy band gap. Moreover, it should be stressed that the UV-Vis absorption edge of TBNTs-1 was slightly larger than that of MTB, but less than that of pure BiOBr nanoplates, which may result from the specific configurations of the TBNTs-1 samples as well as the quantum size-dependent effects.22,25,39–41 As we know, the unique finned-tube structure of the TiO2@BiOBr heterojunctions can reduce the scattering of incident light and display better visible light response ability than that of the MTB sample. Furthermore, the size dimension of BiOBr nanoplates also has a certain influence on the optical property of the samples significantly. As mentioned above, the size of nanoplates in TBNTs-1 sample was smaller than that of pure BiOBr. Thus, due to the size quantization effects,10,20,42 a slight blue-shift absorption of TBNTs-1 sample can be observed in comparison with pure BiOBr nanoplates. On the other hand, the BiOBr nanoplates with 2D nanostructures would be beneficial for the entry and multi-reflection of the visible light, which can contribute to the increased effective optical path length of photons and higher light harvesting efficiency.
 | ||
| Fig. 6 (A) UV-vis diffuse reflection spectra and (B) plots of (αhν)1/2 versus the photon energy (hν) for all samples. (a) Pure BiOBr nanoplates. (b) TBNTs-1. (c) MTB. (d) Sulfated TiO2 NTs. (e) P25 TiO2. | ||
For a crystalline semiconductor, the Eg values of the as-prepared samples can be calculated by the Kubelka–Munk formula (eqn (S1)†).43,44 As depicted in Fig. 6B, the modified TiO2 NTs and P25TiO2 samples exhibited strong photo absorption in the UV region with the corresponding band gap energy at 3.17 and 3.13 eV, respectively, which were well consistent with the relevant reported values.9,45 Meanwhile, the band gaps of pure BiOBr nanoplates, TBNTs-1 and MTB samples were calculated to be 2.62, 2.65 and 2.70 eV, respectively, which were narrower than the theoretical values of BiOBr and TiO2,37,46,47 indicating that these samples can be excited by visible light. Additionally, the positions of the conduction band (CB) and valence band (VB) edges at the point of zero charge (pHZPC) were also estimated according to the atom's Mulliken electronegativity equation (eqn (S2)†).48,49 Thus, the CB and VB potentials of BiOBr existed in TBNTs-1 were estimated to be +0.36 eV and +2.98 eV, separately. Meanwhile, the corresponding ECB and EVB of the modified TiO2 NTs also can be determined to be −0.48 eV and +2.69 eV, respectively. It is no doubt that the two substances had well-matched band structures which can stagger with each other. Namely, the as-formed TiO2@BiOBr heterojunctions classified as the type II heterostructure would benefit the interfacial charge transfer and restrain the recombination of photo-generated electron–hole pairs, subsequently, resulting in the enhanced photocatalytic activity.3,9,33,50
Moreover, the recombination rate of the photo-generated charge carriers was estimated by photoluminescence (PL) spectroscopy analysis.51,52 As shown in Fig. 7a, among all the as-prepared samples, the TBNTs-1 sample displayed the lowest intensity of the emission peak at around 448 nm, indicating that the recombination rate of the photo-generated electron–hole pairs could be effectively suppressed in this sample.
 | ||
| Fig. 7 (a) PL spectra of all samples excited at the light wavelength of 325 nm; (b) the transient photocurrents density response of different photocatalysts electrodes with light on–off cycles under visible light irradiation (≥420 nm); (c) EIS Nyquist plots of different electrodes; (d) the local EIS Nyquist plots of different electrodes. (1) P25 TiO2; (2) sulfated TiO2 NTs; (3) pure BiOBr nanoplates; (4) MTB; (5) TBNTs-1. | ||
As shown in Fig. 7b, the photo-generated charge separation efficiency of as-fabricated samples are further confirmed by the transient photocurrent density responses in an on–off cycle mode. Under visible light irradiation, the TBNTs-1 sample showed the notable enhancement of the photocurrent density response in comparison with sulfated TiO2 NTs, P25 TiO2, MTB and pure BiOBr nanoplate samples. It is believed that the stronger photocurrent intensity can be attributed to the more efficient separation of photo-generated electron–hole pairs.53–56 Furthermore, when the irradiation was interrupted, obvious residual current still can be observed in TBNTs-1 sample and the slower decay of the photocurrent implied that the lifetime of the charge carriers are prolonged. Based on the aformentioned results, it can be inferred that the construction of the finned-tube structured TiO2@BiOBr heterojunctions could facilitate photo-induced charge transfer and restrain the recombination of photo-generated electron–hole pairs efficiently. Electrochemical impedance spectroscopy (EIS) was also employed to investigate the photo-generated charge separation process. As can be seen in Fig. 7c, all the impedance spectra were similar and only the semicircle part at high frequency region could be observed on the EIS Nyquist plots. The semicircle in the high-frequency region corresponds to the charge-transfer process and the arc radius reflects the resistance (Rct) of the charge transfer at the solid/liquid interface.57–61 Compared with sulfated TiO2 NTs and P25 TiO2, the radius of semicircle over TBNTs-1 electrode was decreased, suggesting a drastic decrease in the resistance and enhanced photo-induced electrons and holes separation. The Rct of the pure BiOBr nanoplates (Fig. 7d) and MTB electrodes was lower than that in the TBNTs-1 electrode, which is attributed to the existence of abundant ultrathin BiOBr nanoplates with high electrical conductivity. As a consequence, the results of photoelectrochemical measurements indicated that the intimately interfacial junction can significantly improve the separation of photo-generated electron–hole pairs and reduce the interfacial electron-transfer resistance.
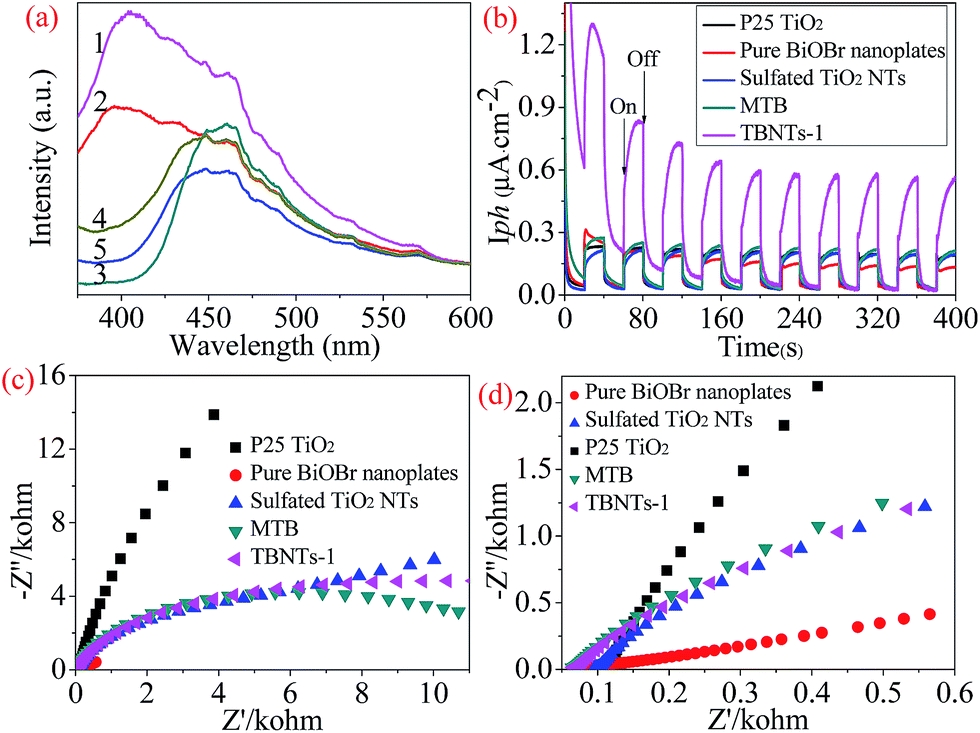
As shown in Fig. 8a, the photocatalytic performance of the resultant samples were evaluated by the degradation of Rhodamine B (RhB, 10 mg L−1) solution under visible light irradiation (λ ≥ 420 nm). For comparison, the photocatalytic performance of the modified TiO2 NTs, commercial P25TiO2, a blank experiment (without any photocatalyst) as well as the control test in the dark were also investigated under the same conditions. As mentioned above, P25 TiO2 and the modified TiO2 NTs could not be excited by the visible light due to the large band gaps, implying the negligible visible-light photocatalytic activity. It can be seen that the concentration of RhB has a slightly change with the increase of irradiation time both in the absence of photocatalyst (4.8%) and in the presence of the modified TiO2 NTs (4.1%) after 24 min irradiation, respectively, which was ascribed to the photosensitization mechanism of RhB.37 Interestingly, the degradation efficiency of commercial P25 TiO2 could reach to 15.6% which was slightly higher than that of TiO2 NTs mentioned above. The causes of this phenomenon can be interpreted as that the coexistence and optimal ratio of rutile and anatase TiO2 in commercial P25 TiO2 will facilitate the photosensitization effect.13,62 In addition, the MTB and pure BiOBr samples showed enhanced photocatalytic performance and approximately 82.9% and 84.5% of RhB were removed under visible light irradiation for 24 min, respectively, attributing to the existent BiOBr with visible light response ability. Notably, no obvious change in the concentration of RhB in the presence of TBNTs-1 sample under dark condition, suggesting that TBNTs-1 sample have reached adsorption–desorption equilibrium. However, the as-prepared TBNTs-1 exhibited the best visible light photocatalytic activity and more than 97% of RhB was degraded under visible light irradiation for 24 min. As far as we know, the photocatalytic performance of the reported congeneric catalysts cannot be mentioned in the same breath with that of TBNTs-1 sample. This comparison suggests that the synthesized TiO2@BiOBr heterojunctions could significantly enhance the photocatalytic performance. As mentioned previously, the geometry of TiO2@BiOBr hybrids considerably affects the resulting degradation efficiency. The as-prepared finned-tube structured TiO2@BiOBr heterojunctions possessed a large length-diameter ratio and thus induced specific surface area which not only displays enhanced adsorption ability of RhB molecules (Fig. S6†), but also can provide a great deal of surface active sites. Furthermore, the cavity of TiO2 NTs and the lamellar structural BiOBr nanoplates were beneficial for the diffusion of contamination and light multi-reflection resulting in a high light harvesting efficiency.63 Moreover, the TiO2@BiOBr heterojunctions with the staggered gap have greatly promoted the interfacial charge transfer and effectively inhibited the recombination of photo-generated electron–hole pairs.64 Consequently, all these aspects contributed to the comprehensively improved photocatalytic performance.
 | ||
| Fig. 8 (a) Plots of the decomposition of RhB solution concentration versus irradiation time by different samples and (b) recycle runs in the photocatalytic degradation of RhB by TBNTs-1 sample. | ||
As depicted in Fig. 8b, the TBNTs-1 sample displayed sustainable photocatalytic performance and the degradation efficiency remained at 95.5% after seven cycles for the photodecomposition of RhB. The results demonstrate that the TBNTs-1 sample exhibits high stability and superior photocatalytic activity which can be a promising photocatalyst for degradation of organic contaminants and other applications.
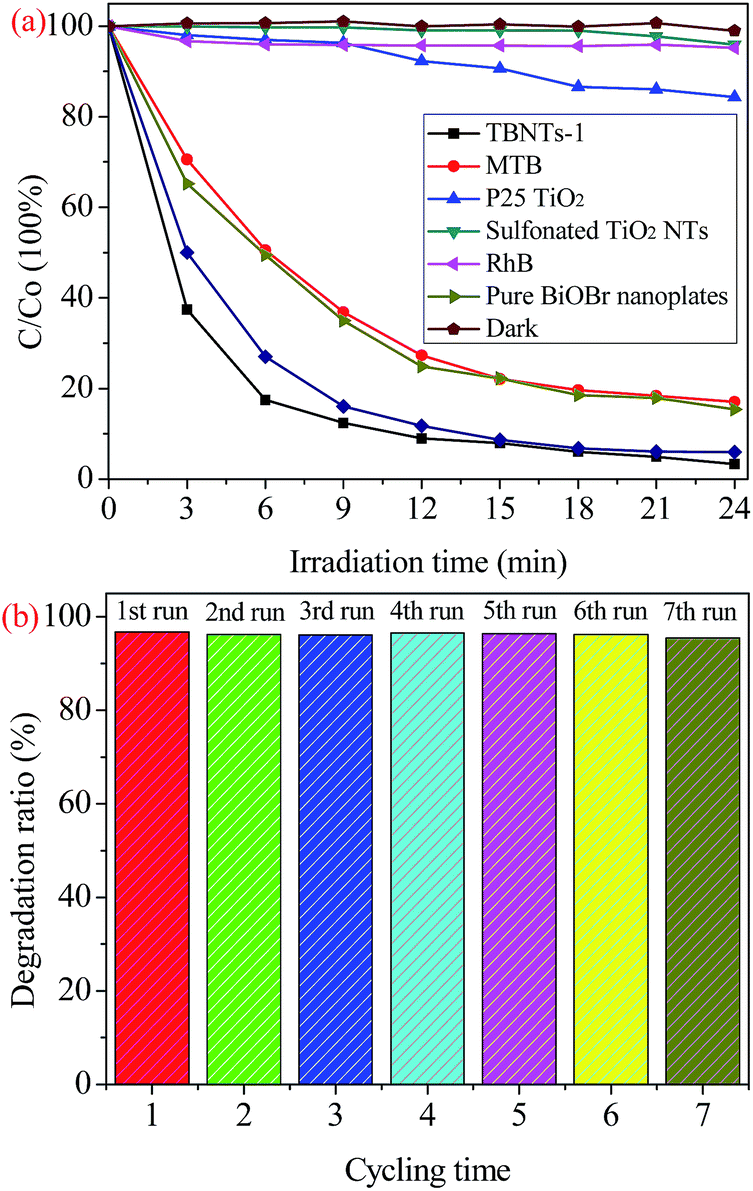
To ascertain the main reactive species involved in the photocatalytic reaction, the radicals and holes trapping experiments were designed by adding various scavengers to the TBNTs-1 sample. In this study, different scavenges of ethylenediaminetetraacetic acid disodium (EDTA-2Na), isopropyl alcohol (IPA) and benzoquinone (BQ) were introduced to scavenge the holes (h+), hydroxyl radicals (·OH), and superoxide radical anions (·O2−) species in the photodegradation of RhB, respectively. The dosages referred to previous research.65–67 As depicted in Fig. 9a, compared with no scavenger, the degradation efficiency of RhB was significantly inhibited with the addition of BQ (10 mM) and EDTA-2Na (10 mM), whereas IPA (1 mM) showed little effect on the degradation of organic contaminant. These results elucidate that both ·O2− and h+ should be the main reactive species in the photocatalytic degaradtion of RhB by the TiO2@BiOBr heterojunction under visible light irradiation.
 | ||
| Fig. 9 (a) Plots of photodegradation of RhB over TBNTs-1 photocatalyst with different scavengers under visible light irradiation. (b) Scheme illustrating the principle of charge-transfer and the photocatalytic processes on the interface of the TBNTs-1 sample under visible light irradiation. | ||
Based on the above analysis and discussion, a possible mechanism to explain the superior photocatalytic activity of TBNTs-1 sample was also proposed, as schematically illustrated in Fig. 9b. There are two possible pathways which lead to the rapid photodegradation process of organic dyes: (1) the intrinsically photocatalytic reaction. During photocatalytic degradation, RhB molecules were firstly adsorbed onto the surface of the finned-tube structured TiO2@BiOBr heterojunctions. Under visible light irradiation, TiO2 cannot be excited due to its intrinsic wide band gap energy, while it is just the opposite that BiOBr with narrower band gap is easily excited and produce photo-induced electron–hole pairs and the generated electrons subsequently transfer to the CB, leaving holes at the valence band of BiOBr. It is generally accepted that photo-induced charge carriers would be effectively separated and transfer from the bulk to the surface at a short distance due to the strong internal static electric field induced in the tiny nanoplates. On the one hand, the photo-generated holes have stronger oxidation ability which could oxidize the pollutants adsorbed on the photocatalyst surface directly and improve the photocatalytic activity. On the other hand, the intimately interfacial contact between BiOBr nanoplates and TiO2 NTs in coupling system is favorable for effective separation and smooth transfer of photo-induced charge. According to the previous band gap calculations, the corresponding CB and VB positions of TiO2 are both more negative than those of BiOBr which offer the opportunity to construct the straddling gap in the heterojunctions. In this case, photo-generated holes are prone to migrate from the VB of BiOBr to that of TiO2, while the same mobility cannot happen in photo-generated electrons at the CB of BiOBr. This transfer process can reduce the recombination of charges effectively. Of course, the holes accumulated in the VB of TiO2 sever as main active species responsible for RhB photodegradation which can directly oxidize the organic contaminants into their end products.5,11,33,50
(2) Photo-excitation and photosensitization of RhB. The photosensitization mechanism is another factor to influence the photodegradation of RhB. Owing to the photosensitization effect, the dye RhB were excited to form its excited state (RhB*) under visible light irradiation and the higher LUMO energy level of the dye is beneficial for the efficient injection of excited electrons into the CB of BiOBr and TiO2, simultaneously.11,40 Meanwhile, the excited state of RhB was converted to the cationic dye radical (RhB˙+). It is understandable that electrons on CB of TiO2 quickly move into CB of BiOBr due to the more positive CB potential of BiOBr. Finally, the electrons in the CB of BiOBr could easily react with oxygen molecules absorbed on the surface of TiO2@BiOBr heterojunctions or dissolved in water to produce superoxide radical anions (·O2−) and hydrogen peroxide (H2O2). As a result, the existent cationic dye radical (RhB˙+) can be directly reduced by the ·O2−, transformed and decomposed into intermediate products and eventually converted to CO2 and H2O.42,68–70 Therefore, the combinations of these active ingredients result in the significant enhancement of photocatalytic performance for visible light-driven photocatalyst.
4 Conclusions
In this article, we have demonstrated a novel and simple method to fabricate the finned-tube structured TiO2@BiOBr heterojunctions with enhanced photocatalytic performance. The synergetic effect on the intrinsic nature of ultrathin BiOBr nanoplates and the unique structure of 1D TiO2 nanotubes as well as the intimately interfacial junction not only resulting in significant improvements in photon acquisition probability and the expansion of the light absorption edge, but also can greatly increase the transport efficiency of photo-generated charge carriers and facilitate the efficient separation of photo-generated electron–hole pairs. These factors lead to extraordinary visible-light photocatalytic performance, robust structural stability and outstanding cycle life of as-prepared photocatalyst. This work provides a new avenue for the controllable synthesis of intriguing hybrid nanoarchitecture with tiny 2D nanoplates on 1D nanotubes which exhibits improved photocatalytic activity. This coupling photocatalyst can be synthesized on a large scale via the simple solvothermal process and make it feasible for industrial application in disposal of water pollution and photocatalytic water splitting as well as other functional solar energy conversion devices.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (Grant No. 21303130); the Natural Science Basic Research Plan in Shaanxi Province of China (Grant No. 2014JQ2066) and the Fundamental Research Funds for the Central Universities. The TEM and SEM work were done at International Center for Dielectric Research (ICDR), Xi'an Jiaotong University, Xi'an, China; the authors also thank Ms Dai and Mr Ma for their help in using SEM, EDX and TEM, respectively.Notes and references
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- L. Ruan, J. Liu, Q. Zhou, J. Hu, G. Xu, X. Shu and Y. Wu, New J. Chem., 2014, 38, 3022–3028 RSC.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- J. Chen, M. Guan, W. Cai, J. Guo, C. Xiao and G. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 20909–20914 RSC.
- G. Jiang, R. Wang, X. Wang, X. Xi, R. Hu, Y. Zhou, S. Wang, T. Wang and W. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 4440–4444 CAS.
- W. Lin, X. Wang, Y. Wang, J. Zhang, Z. Lin, B. Zhang and F. Huang, Chem. Commun., 2014 10.1039/c3cc41498a.
- Z. Y. Zhao and W. W. Dai, Inorg. Chem., 2014, 53, 13001–13011 CrossRef CAS PubMed.
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC.
- X. Cao, Z. Lu, L. Zhu, L. Yang, L. Gu, L. Cai and J. Chen, Nanoscale, 2014, 6, 1434–1444 RSC.
- X. X. Wei, H. Cui, S. Guo, L. Zhao and W. Li, J. Hazard. Mater., 2013, 263, 650–658 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, C. Fan, X. Wang, X. Hao and Y. Chi, J. Solid State Chem., 2013, 199, 224–229 CrossRef CAS.
- X. Li, G. Jiang, Z. Wei, X. Wang, W. Chen and L. Shen, MRS Commun., 2013, 3, 219–224 CrossRef CAS.
- X. Xu, G. Yang, J. Liang, S. Ding, C. Tang, H. Yang, W. Yan, G. Yang and D. Yu, J. Mater. Chem. A, 2014, 2, 116–122 CAS.
- Y. Tang, Y. Zhang, J. Deng, J. Wei, H. L. Tam, B. K. Chandran, Z. Dong, Z. Chen and X. Chen, Adv. Mater., 2014, 26, 6111–6118 CrossRef CAS PubMed.
- B. Weng, S. Liu, Z. R. Tang and Y. J. Xu, RSC Adv., 2014, 4, 12685–12700 RSC.
- W. Ni, F. Liang, J. Liu, X. Qu, C. Zhang, J. Li, Q. Wang and Z. Yang, Chem. Commun., 2011, 47, 4727–4729 RSC.
- Y. Tang, L. Liu, X. Wang, H. Zhou and D. Jia, RSC Adv., 2014, 4, 44852–44857 RSC.
- H. Zhou, L. Liu, X. Wang, F. Liang, S. Bao, D. Lv, Y. Tang and D. Jia, J. Mater. Chem. A, 2013, 1, 8525–8528 CAS.
- X. Xu, H. Tan, K. Xi, S. Ding, D. Yu, S. Cheng, G. Yang, X. Peng, A. Fakeeh and R. V. Kumar, Carbon, 2015, 84, 491–499 CrossRef CAS.
- Y. Huo, J. Zhang, M. Miao and Y. Jin, Appl. Catal., B, 2012, 111, 334–341 CrossRef.
- A. Dash, S. Sarkar, V. N. Adusumalli and V. Mahalingam, Langmuir, 2014, 30, 1401–1409 CrossRef CAS PubMed.
- Z. Deng, D. Chen, B. Peng and F. Tang, Cryst. Growth Des., 2008, 8, 2995–3003 CAS.
- D. Zhang, J. Li, Q. Wang and Q. Wu, J. Mater. Chem. A, 2013, 1, 8622–8629 CAS.
- L. Chen, R. Huang, M. Xiong, Q. Yuan, J. He, J. Jia, M. Y. Yao, S. L. Luo, C. T. Au and S. F. Yin, Inorg. Chem., 2013, 52, 11118–11125 CrossRef CAS PubMed.
- S. Shenawi-Khalil, V. Uvarov, S. Fronton, I. Popov and Y. Sasson, J. Phys. Chem. C, 2012, 116, 11004–11012 CAS.
- M. Gao, D. Zhang, X. Pu, H. Li, J. Li, X. Shao and K. Ding, Mater. Lett., 2015, 140, 31–34 CrossRef CAS.
- B. Zhang, G. Ji, M. A. Gondal, Y. Liu, X. Zhang, X. Chang and N. Li, J. Nanopart. Res., 2013, 15, 1–9 Search PubMed.
- J. Shang, W. Hao, X. Lv, T. Wang, X. Wang, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, ACS Catal., 2014, 4, 954–961 CrossRef CAS.
- J. Zhang, F. Shi, J. Lin, D. Chen, J. Gao, Z. Huang, X. Ding and C. Tang, Chem. Mater., 2008, 20, 2937–2941 CrossRef CAS.
- J. Fu, Y. Tian, B. Chang, F. Xi and X. Dong, J. Mater. Chem., 2012, 22, 21159–21166 RSC.
- R. Yuan, T. Chen, E. Fei, J. Lin, Z. Ding, J. Long, Z. Zhang, X. Fu, P. Liu, L. Wu and X. Wang, ACS Catal., 2011, 1, 200–206 CrossRef CAS.
- Y. Tian, B. Chang, J. Lu, J. Fu, F. Xi and X. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 7079–7085 CAS.
- F. Chang, Y. Xie, J. Zhang, J. Chen, C. Li, J. Wang, J. Luo, B. Deng and X. Hu, RSC Adv., 2014, 4, 28519–28528 RSC.
- C. Xu, H. Wu and F. L. Gu, J. Hazard. Mater., 2014, 275, 185–192 CrossRef CAS PubMed.
- Z. Liu, B. Wu, Y. Zhu, D. Yin and L. Wang, Catal. Lett., 2012, 142, 1489–1497 CrossRef CAS.
- X. X. Wei, C. M. Chen, S. Q. Guo, F. Guo, X. M. Li, X. X. Wang, H. T. Cui, L. F. Zhao and W. Li, J. Mater. Chem. A, 2014, 2, 4667–4675 CAS.
- C. Xue, T. Wang, G. Yang, B. Yang and S. Ding, J. Mater. Chem. A, 2014, 2, 7674–7679 CAS.
- G. Yang, B. Yang, T. Xiao and Z. Yan, Appl. Surf. Sci., 2013, 283, 402–410 CrossRef CAS.
- W. Zhang, Q. Zhang and F. Dong, Ind. Eng. Chem. Res., 2013, 52, 6740–6746 CrossRef CAS.
- H. Zhang, Y. Yang, Z. Zhou, Y. Zhao and L. Liu, J. Phys. Chem. C, 2014, 118, 14662–14669 CAS.
- K. Li, H. Zhang, Y. Tang, D. Ying, Y. Xu, Y. Wang and J. Jia, Appl. Catal., B, 2015, 164, 82–91 CrossRef CAS.
- J. Xiong, Q. Dong, T. Wang, Z. Jiao, G. Lu and Y. Bi, RSC Adv., 2013, 4, 583–586 RSC.
- Y. R. Jiang, H. P. Lin, W. H. Chung, Y. M. Dai, W. Y. Lin and C. C. Chen, J. Hazard. Mater., 2015, 283, 787–805 CrossRef CAS PubMed.
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC.
- H. Tian, J. Li, M. Ge, Y. Zhao and L. Liu, Catal. Sci. Technol., 2012, 2, 2351–2355 CAS.
- Y. X. Yu, W. X. Ouyang and W. D. Zhang, J. Solid State Electrochem., 2014, 18, 1743–1750 CrossRef CAS.
- F. de Angelis, C. Di Valentin, S. Fantacci, A. Vittadini and A. Selloni, Chem. Rev., 2014, 114, 9708–9753 CrossRef CAS PubMed.
- J. Di, J. Xia, Y. Ge, L. Xu, H. Xu, J. Chen, M. He and H. Li, Dalton Trans., 2014, 43, 15429–15438 RSC.
- H. Lin, J. Cao, B. Luo, B. Xu and S. Chen, Chin. Sci. Bull., 2012, 57, 2901–2907 CrossRef CAS.
- W. Zhang, Y. Sun, F. Dong, W. Zhang, S. Duan and Q. Zhang, Dalton Trans., 2014, 43, 12026–12036 RSC.
- S. Kumar, S. Khanchandani, M. Thirumal and A. K. Ganguli, ACS Appl. Mater. Interfaces, 2014, 6, 13221–13233 CAS.
- D. Jiang, J. Li, C. Xing, Z. Zhang, S. Meng and M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 19234–19242 CAS.
- W. Zhao, J. Li, Z. Wei, S. Wang, H. He, C. Sun and S. Yang, Appl. Catal., B, 2015, 179, 9–20 CrossRef CAS.
- W. Lu, T. Xu, Y. Wang, H. Hu, N. Li, X. Jiang and W. Chen, Appl. Catal., B, 2016, 180, 20–28 CrossRef CAS.
- Y. Hong, Y. Jiang, C. Li, W. Fan, X. Yan, M. Yan and W. Shi, Appl. Catal., B, 2016, 180, 663–673 CrossRef CAS.
- Z. Pei, S. Weng and P. Liu, Appl. Catal., B, 2016, 180, 463–470 CrossRef CAS.
- F. Liang and Y. Zhu, Appl. Catal., B, 2016, 180, 324–329 CAS.
- P. Zhang, L. Wang, X. Zhang, C. Shao, J. Hu and G. Shao, Appl. Catal., B, 2015, 166–167, 193–201 CAS.
- Z. Chen, H. Jiang, W. Jin and C. Shi, Appl. Catal., B, 2016, 180, 698–706 CrossRef CAS.
- J. Han, Z. Liu, K. Guo, X. Zhang, T. Hong and B. Wang, Appl. Catal., B, 2015, 179, 61–68 CrossRef CAS.
- Z. Fan, J. Liang, W. Yu, S. Ding, S. Cheng, G. Yang, Y. Wang, Y. Xia, K. Xi and R. V. Kumarb, Nano Energy, 2015, 16, 152–162 CrossRef CAS.
- J. ChuláKim, Y. BokáLee, J. HoonáHong, J. InáLee, J. WookáYang, W. InáLee and N. HwiáHur, Chem. Commun., 2006, 48, 5024–5026 Search PubMed.
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385–9454 CrossRef CAS PubMed.
- C. Xue, J. Xia, T. Wang, S. Zhao, G. Yang, B. Yang, Y. Dai and G. Yang, Mater. Lett., 2014, 133, 274–277 CrossRef CAS.
- W. K. Jo and T. Sivakumar Natarajan, ACS Appl. Mater. Interfaces, 2015, 7, 17138–17154 CAS.
- B. Luo, D. Xu, D. Li, G. Wu, M. Wu, W. Shi and M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 17061–17069 CAS.
- B. Lin, C. Xue, X. Yan, G. Yang, G. Yang and B. Yang, Appl. Surf. Sci., 2015, 357, 346–355 CrossRef CAS.
- L. Ye, J. Liu, C. Gong, L. Tian, T. Peng and L. Zan, ACS Catal., 2012, 2, 1677–1683 CrossRef CAS.
- L. Kong, Z. Jiang, H. H. Lai, R. J. Nicholls, T. Xiao, M. O. Jones and P. P. Edwards, J. Catal., 2012, 293, 116–125 CrossRef CAS.
- T. Wang, X. Yan, S. Zhao, B. Lin, C. Xue, G. Yang, S. Ding, B. Yang, C. Ma, G. Yang and G. Yang, J. Mater. Chem. A, 2014, 2, 15611–15619 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20510g |
| This journal is © The Royal Society of Chemistry 2015 |