
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trivalent Gd-DOTA reagents for modification of proteins†
Martin J. Fisherab,
Daniel J. Williamsonab,
George M. Burslemab,
Jeffrey P. Planteab,
Iain W. Manfieldbc,
Christian Tiedebc,
James R. Aultbc,
Peter G. Stockleybc,
Sven Pleinde,
Azhar Maqboolde,
Darren C. Tomlinsonbc,
Richard Fosterabe,
Stuart L. Warrinerab and
Robin S. Bon*abde
aSchool of Chemistry, University of Leeds, LS2 9JT, UK. E-mail: r.bon@leeds.ac.uk
bAstbury Centre for Structural Molecular Biology, University of Leeds, UK
cSchool of Molecular and Cellular Biology, University of Leeds, UK
dLeeds Institute of Cardiovascular and Metabolic Medicine, University of Leeds, UK
eMultidisciplinary Cardiovascular Research Centre, University of Leeds, UK
First published on 2nd November 2015
Abstract
The development of novel protein-targeted MRI contrast agents crucially depends on the ability to derivatise suitable targeting moieties with a high payload of relaxation enhancer (e.g., gadolinium(III) complexes such as Gd-DOTA), without losing affinity for the target proteins. Here, we report robust synthetic procedures for the preparation of trivalent Gd-DOTA reagents with various chemical handles for site-specific modification of biomolecules. The reagents were shown to successfully label proteins through isothiocyanate ligation or through site-specific thiol–maleimide ligation and strain-promoted azide–alkyne cycloaddition.
Introduction
Magnetic resonance imaging (MRI) is widely used for non-invasive imaging of physiological processes in both clinical medicine and pre-clinical research. MRI is especially advantageous because, in comparison to other imaging modalities, it offers the opportunity to image deep into tissue without the need for ionising radiation, and because it provides 3D images with sub-millimeter spatial resolution.1–3 Most MRI methods rely on 1H NMR signals of water protons, so signals depend on their concentrations and relaxation times (T1 and T2). Chemical contrast agents, for example those based on gadolinium(III) or manganese(II) complexes or on iron oxide nanoparticles, can significantly enhance the sensitivity of MRI, and have already had major impact on (pre)clinical imaging. All clinically approved MRI contrast agents that are currently in use are based on the paramagnetic Gd3+ ion,4 which can shorten the spin–lattice relaxation time (T1) of water in its coordination spheres. Efficient Gd3+-based MRI contrast agents undergo rapid exchange of inner-sphere water molecules with bulk water, resulting in significant enhancement of MR contrast by low micromolar concentrations of Gd3+.1–3Targeted MRI contrast agents consist of a targeting moiety and one or more gadolinium complexes. The targeting moiety is usually a small molecule or peptide. These reagents can be used to localise specific proteins – typically proteins present in the blood or extracellular matrix, or extracellular domains of membrane proteins – in vivo with high spatial resolution. Examples are MRI contrast agents targeted to serum albumin, collagen, fibrin,2,3 and various abundant receptors (e.g., progesterone, folate, dopamine glutamate).1 In addition, a contrast agent based on CTB (cholera toxin subunit B) has been used to successfully image neuronal connections in vivo.5
The development of protein-targeted MRI contrast agents crucially depends on the availability of suitable protein-targeting moieties that can be labelled efficiently with (multiple) gadolinium complexes while preserving: (1) affinity of the targeting moiety for its protein target; (2) strong binding of toxic Gd3+ to its chelating ligand; (3) relaxivity of the gadolinium complex; (4) rapid tissue penetration/target binding and clearance of unbound reagents; and (5) clearance of all reagent from the body before metabolism-related release of Gd3+.
For many proteins, no suitable small molecule ligands are available, and the use of biomolecules as targeting moieties would be desirable. For example, antibody- and affibody-based MRI contrast agents have been developed that enable the targeted imaging of EGFR and Her-2,6 including a two-component approach based on a biotinylated antibody and Gd-DTPA-labelled avidin to increase the gadolinium payload (with a detection limit of ca. 106 receptors per cell).7 Affibodies and other antibody mimetics have significant advantages over antibodies as targeting moieties: they are smaller, causing rapid tissue penetration and blood clearance; they can be selected rapidly in vitro against a wide range of biomolecules; they are easier to produce in homogeneous batches that are devoid of post-translational modifications; and they can be labelled site-specifically with suitable chemicals.
Macrocyclic ligands such as DOTA and DO3A are metabolically more stable than linear ones such as DTPA, and they bind Gd3+ much more tightly (pKD = 28 for DOTA vs. 22 for DTPA),8 minimising potential toxicity issues. DOTA can be linked to targeting moieties through amide bond formation with one (or more) of its carboxylic acids. However, the change of a coordinating carboxylate to an amide slightly reduces the binding constant of the Gd-DOTA complex. More importantly, this substitution significantly slows the water exchange rate of the complex (a limiting factor when imaging slow-tumbling molecules).1,3 Our aim was to generate a toolbox of trivalent Gd-DOTA reagents that already contain Gd3+ and can be used to selectively label any targeting moiety (i.e. small molecules, peptides and biomolecules) in one simple step. The trivalent Gd-DOTA reagents were based on DOTA-GA in combination with a diethylenetriamine linker (Fig. 1). DOTA-GA contains a glutamic acid side chain that allows chemical modification without negatively affecting the water exchange rate of the Gd-DOTA complex. In addition, the limited rotational freedom of the short triamine linker would ensure minimal loss of relaxivity through rotational motion (a limiting factor when using clinically prevalent mid-range scanners).1,3
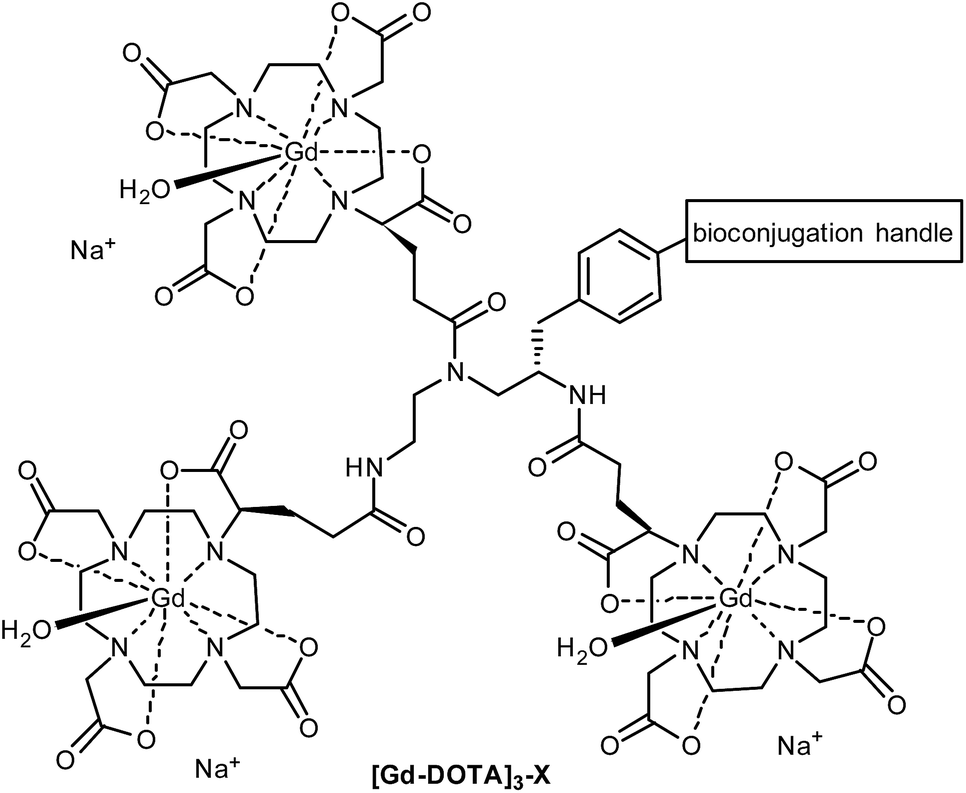 | ||
| Fig. 1 Generic structure of trivalent Gd-DOTA reagents [Gd-DOTA]3-X described in this study (X = reactive handle for chemoselective conjugation to biomolecules). | ||
One trivalent complex [Gd-DOTA]3-X (Fig. 1, X = NCS; compound 14) has previously appeared in the patent literature.9 However, DOTA chemistry is notoriously sensitive to impurities and, in our experience, the details disclosed in this patent are insufficient to allow straight-forward preparation of trivalent Gd-DOTA reagents. Here, we provide detailed (optimised) procedures for the synthesis and purification of one known and two new trivalent Gd-DOTA reagents [Gd-DOTA]3-X. In addition, we report on bioconjugation studies for the modification of proteins such as antibody mimetics.
Results and discussion
Synthesis of (R)-tBu4-DOTA-GA
DOTA-based building block (R)-tBu4-DOTA-GA (6) was synthesised through adaptation of a literature procedure reported to give 6 in high purity and >97% enantiomeric excess (Scheme 1).10 L-Glutamic acid was converted to lactone 1 through diazotisation followed by tert-butyl ester formation, in moderate yield (over 3 steps, after crystallisation).11 Lactone ring opening with 1 equivalent of KOH, followed by benzylation of the intermediate potassium carboxylate, gave alcohol 2, which was mesylated in good yield. Alcohol 2 was easy to store. Therefore, mesylate 3 was always prepared freshly before use.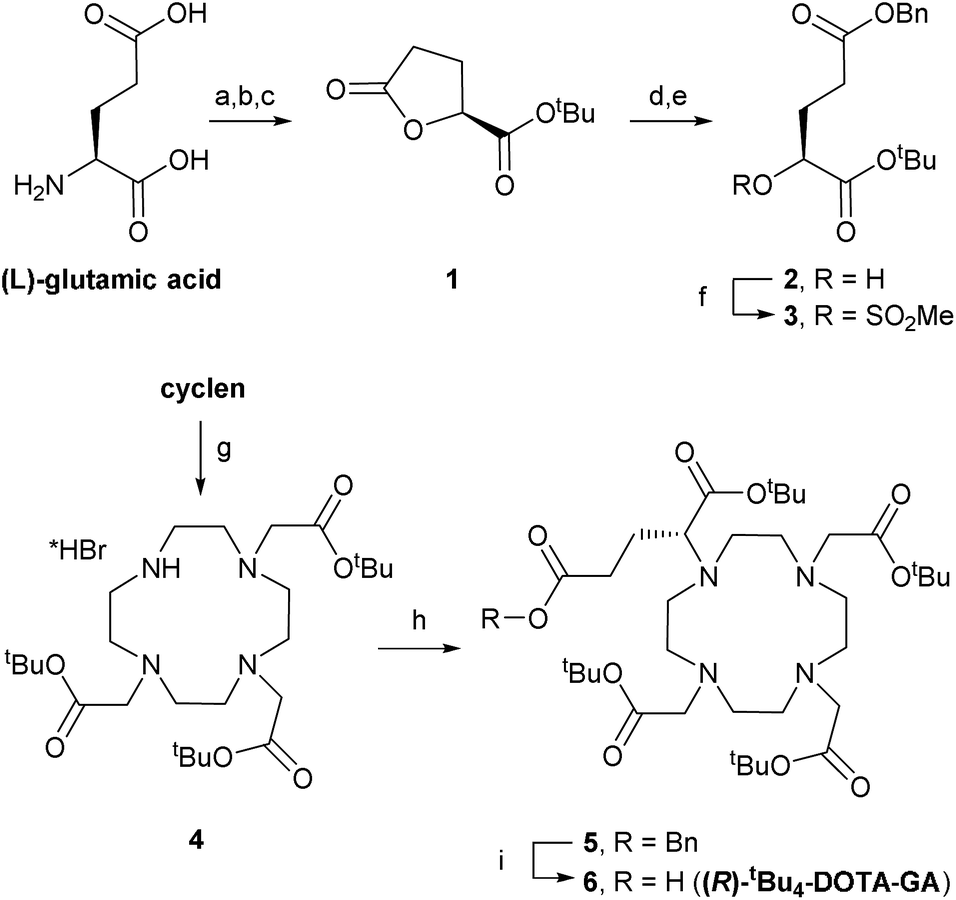 | ||
| Scheme 1 Synthesis of (R)-tBu4-DOTA-GA (6). Reagents and conditions: (a) NaNO2, aq. HCl, dioxane, 0 °C → rt, 1.5 h; (b) (COCl)2, DMF, DCM, 0 °C → rt, 2 h; (c) tBuOH, 2,6-lutidine, DCM, 0 °C → rt, 16 h, 38% over 3 steps; (d) 1 M KOH (1 equiv.), THF, rt, 2 h; (e) benzyl bromide, DMF, 8 h, 55% over 2 steps; (f) MsCl, DIPEA, DCM, 0 °C → rt, 30 min, 71%; (g) tert-butylbromoacetate, DMA, NaOAc, 0 °C → rt, 16 h, 70%; (h) 3, K2CO3, MeCN, rt → 60 °C, 16 h, 70%; (i) H2, 5% Pd/C, MeOH, rt, 16 h, 80%. | ||
Levy et al. synthesised benzyl-protected DOTA-GA 5 by mono-alkylation of cyclen with mesylate 3 followed by triple alkylation with tert-butylbromoacetate. Their procedure required 2 equivalents of cyclen (an expensive building) in the first step to avoid over-alkylation, and removal of excess cyclen after the reaction was essential to avoid problems in subsequent steps.10 In contrast, we isolated tri-functionalised cyclen 4 as its pure HBr salt by crystallisation from chloroform/diethyl ether.12 Alkylation of 4 with 1.2 equivalents of 3 under basic conditions afforded 5.
Benzyl ester 5 was highly sensitive to trans-esterification under basic conditions. Even after filtration of the crude reaction mixture, sufficient potassium carbonate was present to give the corresponding methyl ester upon addition of methanol.13 Therefore, 5 was purified by automated reverse phase (RP; C18) chromatography before hydrogenolysis to afford (R)-tBu4-DOTA-GA (6). We also noticed that amide bond formations with 6 are very sensitive to residual alcohol or water; rapid hydrolysis/methanolysis of activated esters of 6 is consistent with the sensitivity of 5 to trans-esterification. Therefore, 6 (1.35 g) was purified by automated RP (C18) chromatography and subsequent lyophilisation. The use of an acid-free eluent during this purification was crucial to avoid formation of salts, and for consistent results with subsequent amide bond formations. The ee of 6 was >97%, as determined by NMR analysis upon formation of amides with either enantiomer of α-methylbenzylamine, according to reported procedures.10
Synthesis of trivalent Gd-DOTA reagents
Trivalent linker 9 was prepared in 2 steps from L-nitrophenylalanine methyl ester 7 according to a literature procedure (Scheme 2).14 Triamine 9 was isolated as its triple HCl salt, which is hygroscopic and needs to be stored under an inert atmosphere for consistent results in subsequent acylation with (R)-tBu4-DOTA-GA (6). After extensive optimisation of reaction conditions, triple acylation of triamine 9 with 6 using HATU and DIPEA afforded [(R)-tBu4-DOTA]3-NO2 (10) in excellent yield. In contrast to reported procedures, only a small excess of 6 (1.1 equiv. per acylation) was required. The best conversion was seen with freshly freeze-dried 6 (purified as described above) and anhydrous DIPEA. Consistent with the patent literature, purification of 10 by flash chromatography was only partially successful, and gave low yield of pure material.9 However, straight-forward size exclusion chromatography (Sephadex LH-20), followed by lyophilisation, gave 10 in high yield.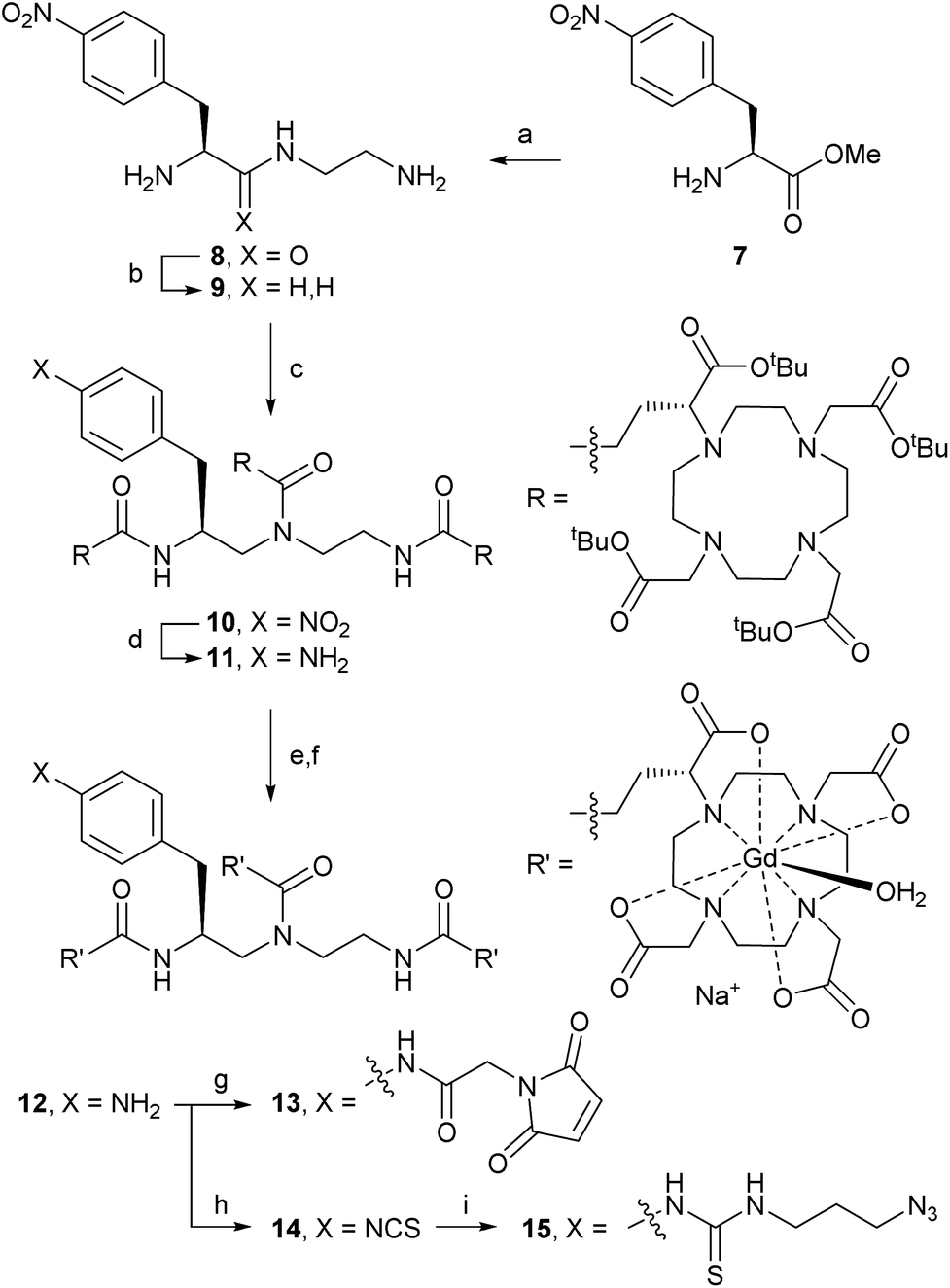 | ||
Scheme 2 Synthesis of the trivalent Gd-DOTA reagents [Gd-DOTA]3-maleimide (13), [Gd-DOTA]3-ITC (14), and [Gd-DOTA]3-N3 (15) via [Gd-DOTA]3-NH2 (12). Reagents and conditions: (a) ethylene diamine, rt, 16 h, 97%; (b) BH3 in THF, 0 °C → reflux, 19 h, 43% (isolated as triple HCl salt); (c) 6, HATU, DIPEA, DMF, 0 °C → rt, 16 h, 88%; (d) H2, 10% Pd/C, MeOH, 40 °C, 16 h, 93%; (e) TFA, TES, MsOH, 0 °C, 4.5 h; (f) GdCl3, aq. NaOH (pH 6.5), rt → 50 °C, 16 h, 75% over 2 steps; (g) 2-maleimidoacetic acid-OSu, HEPES buffer pH 7.3, DMSO, 6 h, 40%; (h) CSCl2, CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1), rt, 16 h, 70%; (i) 3-azidopropylamine, carbonate buffer pH 9.4, DMSO, rt, 2 h, 76%. :H2O (1:1), rt, 16 h, 70%; (i) 3-azidopropylamine, carbonate buffer pH 9.4, DMSO, rt, 2 h, 76%. | ||
Hydrogenation of the nitro group of 10 gave aniline [(R)-tBu4-DOTA]3-NH2 (11) as a pure compound, which was converted into [Gd-DOTA]3-NH2 (12) in two steps. The use of methanesulfonic acid, in addition to TFA and the cation scavenger triethylsilane, was essential for the deprotection of all 12 tert-butyl esters of 11.15 The reaction worked best when kept at or just above 0 °C; significant decomposition and reduced yields were observed if the reaction mixture was allowed to warm up. Therefore, the deprotected intermediate (as its methanesulfonate salt) was separated from the deprotection cocktail by precipitation upon addition of cold diethyl ether. The resulting trivalent DOTA ligand was then directly charged with Gd3+ to afford trivalent DOTA chelate [Gd-DOTA]3-NH2 (12). The crude product 12 was purified by size exclusion chromatography to remove excess salts. Formation of the paramagnetic complex 12 was confirmed by high resolution ESI-MS.15 Purified aniline 12 was prepared in batches of ca. 350–400 mg, and could be stored at −20 °C for >1 year without signs of decomposition.
Next, aniline 12 was modified to install reactive handles for the labelling of targeting moieties, including peptides and biomolecules. Modifications of 12 were performed on small scales (6–50 mg) to produce material for bioconjugation studies. Acylation with the N-hydroxylsuccinimide ester of 2-maleimidoacetic acid (2-maleimidoacetic acid-OSu),16 afforded [Gd-DOTA]3-maleimide (13) suitable for selective modifications of thiols (e.g., cysteines). Treatment of 12 with thiophosgene gave [Gd-DOTA]3-ITC (14) suitable for reactions with nucleophilic amines (e.g., lysines). Isothiocyanate 14 could also be converted into the azide [Gd-DOTA]3-N3 (15), suitable for, for example, copper-catalysed azide–alkyne cycloadditions (CuAAC) or strain-promoted azide–alkyne cycloadditions (SPAAC). 2-Maleimidoacetic acid-OSu and 3-azidopropylamine were separated from trivalent Gd-DOTA reagents 13 and 15, respectively, using size exclusion chromatography before bioconjugation reactions; isothiocyanate 14 did not require purification.
Bioconjugation studies with trivalent Gd-DOTA reagents
Next, the suitability of trivalent Gd-DOTA reagents 13, 14 and 15 for protein modification was investigated. Initially, bioconjugation experiments were performed with lysozyme. Incubation of lysozyme (71 μM in 0.1 M sodium carbonate) with three equivalents of 14 resulted in rapid bioconjugation (Fig. 2a and b). SDS-PAGE analysis showed mainly single and double labelling of lysozyme, and some triple labelling (Fig. 2a, lane 1). The identity of the singly- and doubly-labelled lysozyme was confirmed by ESI-MS (Fig. 2b and the ESI†).17 Preincubation of 14 in 0.1 M sodium carbonate for 45 minutes before addition of lysozyme resulted in identical labelling patterns (Fig. 2a, lane 2), which indicates that 14 is relatively stable in basic aqueous solution. In addition, 14 could be stored as a powder at −20 °C for >1 year without affecting its reactivity.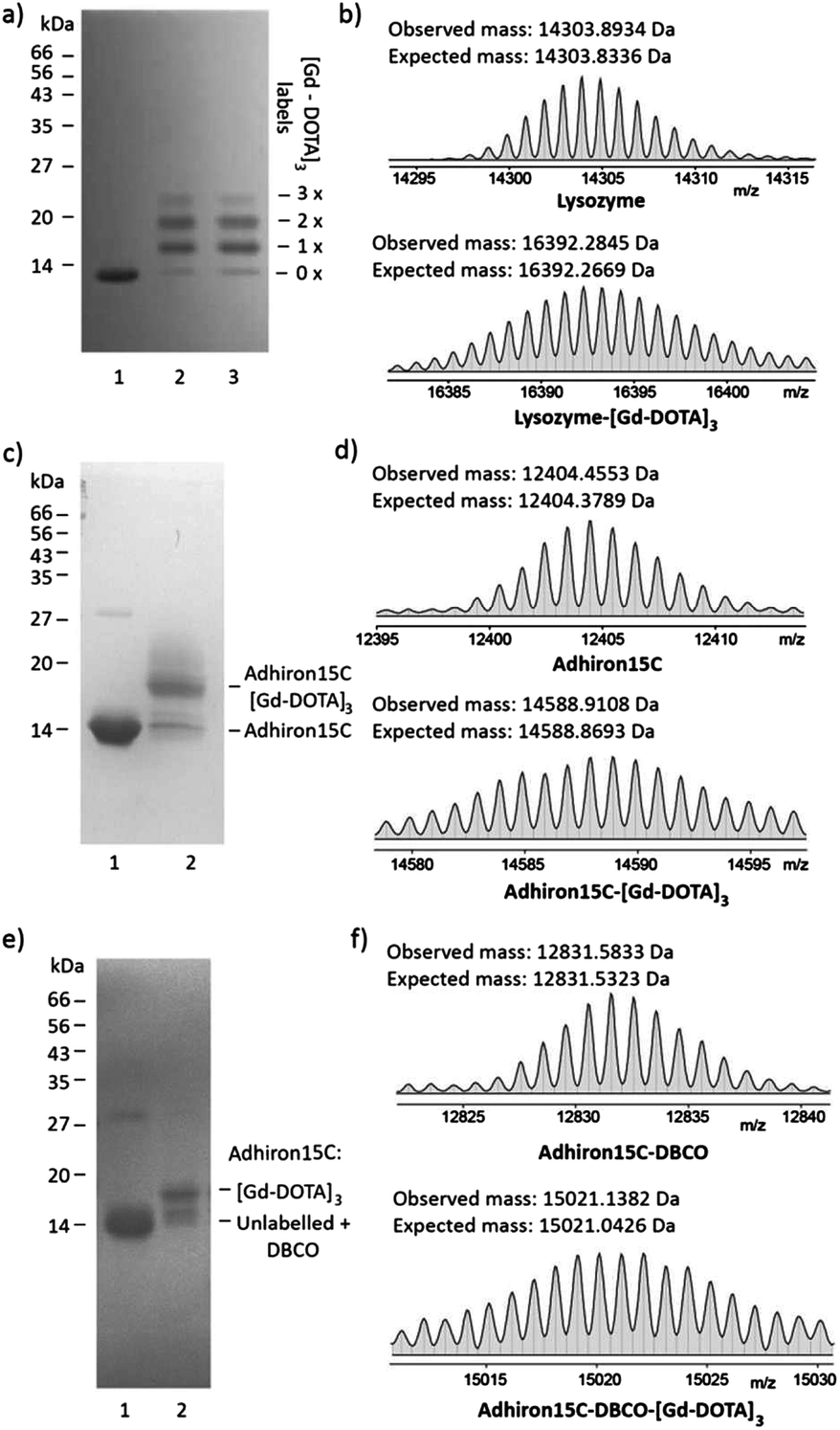 | ||
| Fig. 2 Modification of biomolecules with trivalent Gd-DOTA reagents. (a and b) Reaction of lysozyme (71 μM in 0.1 M Na2CO3) with ITC 14 (3 equiv.; 18 h) resulted in single, double and triple labelling, according to SDS-PAGE (a) and high resolution ESI-MS ((b); deconvoluted spectrum shown) analysis. In (a) lane 1: lysozyme; lane 2: lysozyme after reaction with ITC 14; lane 3: ITC 14 pre-incubated with carbonate buffer for 45 min before reaction with lysozyme. (c and d) Reaction of Adhiron15C (84 μM in phosphate buffer pH 7.4) with maleimide 13 (40 equiv.; 6 h) resulted in single labelling only, according to SDS-PAGE (c) and high resolution ESI-MS ((d); deconvoluted spectrum shown) analysis. In (c) lane 1: Adhiron15C the minor band at higher mass represents a small amount of Adhiron15C dimer (disulfide); lane 2: Adhiron15C upon reaction with maleimide 13. (e and f) Two-step labelling of Adhiron15C (50 μM in phosphate buffer pH 7.4) with azide 15 (2 equiv.; 6 h) resulted in rapid strain-promoted azide–alkyne cycloaddition according to SDS-PAGE (e) and high resolution ESI-MS ((f); deconvoluted spectrum shown) analysis. In (e) lane 1: Adhiron15C upon reaction with dibenzocyclo-octyne maleimide (DBCO-Mal; 20 equiv.; 6 h); lane 2: Adhiron15C-DBCO upon reaction with azide 15 (2 equiv.; 6 h). | ||
Our ongoing research focuses on the use of functionalised Adhirons for targeted imaging. Adhirons (also known as Affimers) are a novel class of small (ca. 12 kDa) antibody mimetics that can be selected rapidly in vitro against a range of targets using phage display protocols.18 Adhirons have excellent binding affinity and specificity, high thermal stability, low production costs and no disulfide bonds. Compared to antibodies, their much smaller size would facilitate more rapid tissue penetration and blood clearance, which is advantageous for in vivo imaging. In addition, the ease of engineering of site-specific modifications, including single or multiple cysteines, makes Adhirons well-suited for site-specific chemical labelling. To test the site-specific modification of Adhirons with trivalent Gd-DOTA reagents, bioconjugation reactions were performed with Adhiron15C, which contains a single cysteine on its C-terminus (pointing away from the binding loops) (Fig. 2c–f). Adhiron15C contains a C-terminal His tag and was purified by Ni nitrilotriacetic acid (NTA) affinity chromatography. Initially, labelling with maleimide 13 was performed directly in the elution buffer, at a protein concentration of 84 μM. SDS-PAGE analysis showed formation of a single labelling product even in the presence of 40 equiv. of 13 (Fig. 2c) and high resolution ESI-MS confirmed the identity of Adhiron15C-[Gd-DOTA]3 (Fig. 2d).
Although the labelling of Adhiron15C with maleimide 13 was successful, a large excess of this precious reagent was needed. To check if inefficient labelling was caused by imidazole and/or tris(2-carboxyethyl)phosphine (TCEP) present in the Adhiron elution buffer, ligation reactions were also attempted after purification of Adhiron15C by dialysis into labelling buffer (PBS containing 10% glycerol and 0.05% Tween-20; pH 7.4) and/or gel filtration. However, this did not significantly improve the labelling reaction, and neither did the use of polymer-supported TCEP (instead of TCEP solution) to reduce disulfide bonds of Adhiron dimers before labelling. Therefore, a more efficient 2-step protocol for Adhiron labelling based on SPAAC19 was developed. Treatment of Adhiron15C (50 μM in labelling buffer) with the commercially available linker dibenzocyclo-octyne maleimide (DBCO-Mal; see the ESI† for structure; 20 equiv.) gave Adhiron15C-DBCO (Fig. 2f).
After buffer exchange (PD-10 column) and concentration, Adhiron15C-DBCO (89 μM in labelling buffer) was treated with azide 15 (2 equiv.), leading to rapid formation of Adhiron15C-DBCO-[Gd-DOTA]3 by SPAAC (Fig. 2e and f). Although conversion was still not complete (most likely because of the sluggish maleimide ligation to form Adhiron15C-DBCO), this procedure gave similar conversion to direct ligation with maleimide 13, but is significantly more efficient in terms of the required amount of trivalent Gd-DOTA reagent.
Conclusions
Trivalent Gd-DOTA reagents have been developed for the site-specific functionalisation of (bio)molecules bearing amines, thiols, and/or (strained) alkynes. Detailed synthetic procedures have been reported, including recommendations regarding purification and storage to assure high-yielding reactions with sensitive intermediates. Procedures for the (site-specific) modification of different proteins were developed. Our reagents allow a range of chemoselective ligation reactions with targeting moieties, including biomolecules. Therefore, they may contribute to the development of novel MRI contrast agents targeted to proteins for which no suitable and/or easy-to-functionalise small molecule- or peptide-based binders are available, but for which phage display protocol can deliver antibody mimetics with high affinity and selectivity. In addition, our reagents could triple the gadolinium payload of previously described CTB-based neuronal tracers.5 We are currently using our reagents to optimise the gadolinium payload on Adhiron-based contrast agents, for example through multiple labelling or the inclusion of a polyvalent scaffold.7Experimental
Synthetic procedures
The following compounds were prepared using literature methods and full reaction details can be found in the ESI:† 1,10 2,10 3,10 4,12 8,14 9,14 2-maleimidoacetic acid (S1)16 and 2-maleimidoacetic acid-OSu (S2).16:H2O (20:80) and purified by automated RP (C18) chromatography using gradient elution (MeCN–H2O–HCOOH, 15:90:0.1 to 85:10:0.1). The eluate was concentrated in vacuo to give an aqueous emulsion (6–10 mL) which was lyophilised to give the title compound (as its double formic acid salt) as a viscous amber oil (2.7 g, 70%). νmax cm−1 2979 and 1725; δH (500 MHz; CDCl3), 9.65–10.15 (br s, 2H), 8.48 (s, 2H), 7.30–7.38 (m, 5H), 5.11 (s, 2H), 3.71–3.79 (m, 4H), 3.44 (s, 2H), 3.34 (dd, J 4.2, 10.1, 1H), 3.12–3.22 (m, 8H), 2.94–3.00 (m, 4H), 2.85–2.94 (m, 4H), 2.38–2.52 (m, 2H), 2.03–2.11 (m, 1H), 1.86–1.94 (m, 1H), 1.44–1.47 (three s, 36H); δC (125 MHz; CDCl3), 172.5, 171.0, 169.4, 168.5, 166.0, 135.7, 128.6, 128.4, 128.3, 82.8, 82.4, 82.3, 66.4, 63.1, 56.4, 55.2, 52.9, 52.5, 49.9, 46.8, 30.9, 28.2, 28.1, 23.7; m/z HRMS (ESI) calcd for C42H71N4O10: 791.5165 [M + H]+, found 791.5174.:H2O (20:80) and purified by automated RP (C18) chromatography using gradient elution (MeCN–H2O, 10:90 to 90:10). The eluate was concentrated in vacuo to give an aqueous solution (5–10 mL) which was lyophilised to give the title compound as a colourless amorphous solid (1.35 g, 80%). νmax cm−1 3426, 3096, 2979, 1954 and 1720; δH (500 MHz; CDCl3) 6.62–6.97 (bs, 1H), 3.59–3.67 (m, 4H), 3.55 (t, J 6.9, 1H), 3.48 (s, 2H), 2.94–3.19 (m, 14H), 2.84–2.92 (m, 2H), 2.51–2.59 (m, 1H), 2.39–2.48 (m, 1H), 1.98–2.06 (m, 1H), 1.87–1.96 (m, 1H), 1.40–1.48 (three s, 36H); δC (125 MHz; CDCl3) 176.1, 171.4, 170.8, 170.3, 81.6, 81.3, 81.1, 63.6, 56.1, 55.7, 52.0, 51.7, 49.4, 33.1, 28.3, 28.2, 27.93, 27.87, 25.3; m/z HRMS (ESI) calcd for C35H65N4O10: 701.4695 [M + H]+, found 701.4702.000 × g, 1 min), and the supernatant was removed and retained before the pellet was redissolved in DMSO (100 μL) and added back to the supernatant. The reaction was spun (Stuart rotator) at room temperature for 4 h before a further aliquot of 2-maleimidoacetic acid-OSu (18 mg, 0.072 mmol) in DMSO (200 μL) was added. The reaction mixture was spun for a further 2 h, then loaded onto a size exclusion chromatography (LH20) column, and the product was eluted with H2O. The eluted product was lyophilised to give the title compound as an off-white solid (20 mg, 40%, some starting material 12 present according to LCMS), which was used immediately for bioconjugation reactions. m/z HRMS (ESI, negative mode) calcd for C74H101Gd3N17O30: 726.4889 M3−, found 726.4918 (complex pattern due to Gd isotopes; the predicted and observed isotopic distributions were identical, see ESI†).:50, 1 mL), and the reaction mixture was stirred vigorously overnight. Once the reaction was complete according to LCMS, the reaction mixture was diluted with water (3 mL) and the aqueous layer was extracted with chloroform (2 × 3 mL). The organic fractions were discarded and the remaining aqueous solution was lyophilised to give the title compound as a pale red amorphous solid (35 mg, 70%). m/z HRMS (ESI, negative mode) calcd for C69H96Gd3N16O27S: 695.1369 M3−, found 695.1346 (complex pattern due to Gd isotopes; the predicted and observed isotopic distributions were identical, see ESI†).Bioconjugation reactions
:1000) into labelling buffer (PBS containing 20% glycerol and 0.05% Tween-20; pH 7.4) to give a protein solution of 57 μM. 6.1 mL of this solution (0.35 μmol) was treated with TCEP in H2O (50 mM; 350 μL; 17.5 μmol), labelling buffer (185 μL) and DBCO-Mal in DMSO (20 mM; 350 μL, 7 μmol) to give a final protein concentration of 50 μM. The reaction was rocked for 6 hours and monitored by mass spectrometry. Upon completion, the material was passed through a buffer exchange column (PD-10, GE Healthcare) according to manufacturer's instructions, eluting 0.5 mL fractions with labelling buffer. Fractions containing protein were identified by BioRad colourimetric assay and pooled. The protein was then concentrated to 89 μM by spin concentrator (3 kDa cut-off), analysed by HRMS, and used immediately in the next step (or flash frozen and stored at −80 °C if required).2 mL of this Adhiron15C-DBCO solution (0.17 μmol) was treated with [Gd-DOTA]3-azide 15 (2 mM in H2O; 175 μL; 0.35 μmol) and the solution was rocked for 6 hours. Upon completion, the material was passed through a buffer exchange column (PD-10, GE Healthcare) according to manufacturer's instructions, eluting 0.5 mL fractions with labelling buffer. Fractions containing protein were identified by BioRad colourimetric assay and pooled. Adhiron15C-DBCO-[Gd-DOTA]3 was concentrated to 323 μM by spin concentrator (3 kDa cut-off), analysed by SDS-PAGE and HRMS, flash frozen and stored at −80 °C.
Acknowledgements
We thank the British Heart Foundation (grant nr NH/12/1/29382) and the Leeds Teaching Hospitals Charitable Foundation (grant nr 3T92/1203) for funding.Notes and references
- N. Sim and D. Parker, Chem. Soc. Rev., 2015, 44, 2122 RSC.
- (a) P. Caravan, Acc. Chem. Res., 2009, 42, 851 CrossRef CAS PubMed; (b) P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- E. Boros, E. M. Gale and P. Caravan, Dalton Trans., 2015, 44, 4804 RSC.
- Mn(II) and iron oxide-based contrast agents have been discontinued, see ref. 2.
- C. W.-H. Wu, O. Vasalatiy, N. Liu, H. Wu, S. Cheal, D.-Y. Chen, A. P. Koretsky, G. L. Griffiths, R. B. H. Tootell and L. G. Ungerleider, Neuron, 2011, 70, 229 CrossRef CAS PubMed ; each CTB subunit was derivatised with 1–6 Gd-DOTA groups using a monovalent Gd-DOTA isothiocyanate.
- J. Qiao, S. Xue, F. Pu, N. White, J. Jiang, Z.-R. Liu and J. J. Yang, J. Biol. Inorg. Chem., 2014, 19, 259 CrossRef CAS PubMed.
- D. Artemov, N. Mori, R. Ravi and Z. M. Bhujwalla, Cancer Res., 2003, 63, 2723 CAS ; on average, the constructs contained 12.5 Gd-DTPA moieties per avidin.
- M. Magerstädt, O. A. Gansow, M. W. Brechbiel, D. Colcher, L. Baltzer, R. H. Knop, M. E. Girton and M. Naegele, Magn. Reson. Med., 1986, 3, 808 CrossRef.
- A. Abujoub, D. R. Buckler, P. D. Caravan, S. Dumas, V. Jacques, S. K. Koerner, A. Kolodziej, D. B. Kumar, R. Looby, A. K. Sato, W.-C. Sun and Z. Zhang, US Pat., PCT/US2006/062760, WO2007/084264 A2, 2007.
- S. G. Levy, V. Jacques, K. L. Zhou, S. Kalogeropoulos, K. Schumacher, J. C. Amedio, J. E. Scherer, S. R. Witowski, R. Lombardy and K. Koppetsch, Org. Process Res. Dev., 2009, 13, 535 CrossRef CAS.
- In the first step of this sequence, a syringe pump was used to add aqueous sodium nitrite solution to the L-glutamate solution; this approach gave significantly higher yields than portion-wise addition of solid sodium nitrite.
- B. Jagadish, G. L. Brickert-Albrecht, G. S. Nichol, E. A. Mash and N. Raghunand, Tetrahedron Lett., 2012, 52, 2058 CrossRef PubMed.
- Unfortunately we did not manage to exploit this reactivity to shorten the synthesis by hydrolysing the ester during workup.
- D. T. Corson and C. F. Meares, Bioconjugate Chem., 2000, 11, 292 CrossRef CAS PubMed.
- To avoid chelation of metal ions other than Gd3+ by the deprotected DOTA derivative, glassware was cleaned with concentrated sulfuric acid before use, HPLC-grade solvents, TFA and MsOH were used for the conversion of 11 → 12. No products incorporating metal ions other than Gd3+ were detected by HRMS.
- T.-P. Wang, N. C. Ko, Y.-C. Su, E.-C. Wang, S. Severance, C.-C. Hwang, Y. T. Shih, M. H. Wu and Y.-H. Chen, Bioconjugate Chem., 2012, 23, 2417 CrossRef CAS PubMed.
- The intensity of ESI-MS peaks resulting from lysozyme-([Gd-DOTA]3)2 was weak (see the ESI†), and minor amounts of lysozyme-([Gd-DOTA]3)3 could not be detected by ESI-MS analysis of reaction mixtures. We suggest that SDS-PAGE analysis better reflects the abundance of different labelling species than ESI-MS.
- C. Tiede, A. A. Tang, S. E. Deacon, U. Mandal, J. E. Nettleship, R. L. Owen, S. E. George, D. J. Harrison, R. J. Owens, D. C. Tomlinson and M. J. McPherson, Protein Eng., Des. Sel., 2014, 27, 145 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. T. Dirks, F. P. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for known compounds; materials and methods for bioconjugation reactions; copies of spectra of new compounds and compounds prepared according to new procedures. See DOI: 10.1039/c5ra20359g |
| This journal is © The Royal Society of Chemistry 2015 |