
Anionic phenoxy-amido rare-earth complexes as efficient catalysts for amidation of aldehydes with amines†
Abstract
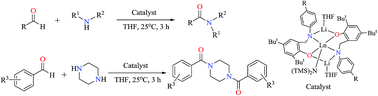
A series of anionic organo-rare-earth amido complexes stabilized by dianionic phenoxy-amido ligands were prepared and their catalytic behavior for amidation reactions of aldehydes with amines was elucidated. Amine elimination reaction of Ln[N(SiMe3)2]3(μ-Cl)Li(THF)3 with an equimolar of lithium aminophenoxy {[HNO]1Li(THF)}2, which was prepared by the reaction of [HNOH]1 {[HNOH]1 = N-p-fluoro-phenyl(2-hydroxy-3,5-di-tert-butyl)benzylamine} with one equivalent of n-BuLi in tetrahydrofuran (THF) in situ, gave the anionic phenoxy-amido rare earth amido complexes [NO]12Ln[N(SiMe3)2][Li(THF)]2 [Ln = Y (1), Yb (2), Sm (3), Nd (4)] in high isolated yields. Similar reactions of Ln[N(SiMe3)2]3(μ-Cl)Li(THF)3 with {[HNO]2Li(THF)}2, and {[HNO]3Li(THF)}2 in THF gave the anionic rare-earth amides [NO]22Ln[N(SiMe3)2][Li(THF)]2 [Ln = Sm (5), Nd (6)] and [NO]32Ln[N(SiMe3)2][Li(THF)]2 [Ln = Sm (7), Nd (8)] {[HNOH]2 = N-p-chloro-phenyl(2-hydroxy-3,5-di-tert-butyl)benzylamine; [HNOH]3 = N-p-bromo-phenyl(2-hydroxy-3,5-di-tert-butyl)benzylamine}, respectively. All of these complexes were fully characterized. X-ray structural determination revealed that these complexes are isostructural, and have solvated monomeric structures. Each of the rare-earth ions is coordinated by two phenoxy-amido ligands and one N(SiMe3)2 group, and the coordination geometry can be described as a distorted trigonal bipyramid. Each of the lithium atoms is surrounded by one aryloxo group, one amido group and one THF molecule, and the coordination geometry can be described as a trigonal plane. The catalytic behavior of these rare-earth amides for the amidation reaction of aldehyde with amine was elucidated. It was found that these complexes are efficient catalysts for this transformation to produce amides in good to excellent yields under mild reaction conditions, and in some cases, diacylamide compounds can be prepared conveniently.
- This article is part of the themed collection: RSC Advances Editors' collection: f Block Chemistry
 Please wait while we load your content...
Please wait while we load your content...

