
Fluorescence amplification detection via terminal protection of small molecule–protein interactions†
Sen Lia,
Yu Lia,
Hongwei Yua,
Zhan Wu*b,
Jianhui Jiangb,
Ruqin Yub and
Yuansheng Wang*a
aCollege of Science, The Naval University of Engineering, Wuhan, 430000, P. R. China. E-mail: qianxun8965@163.com; Fax: +86-027-68771774; Tel: +86-027-68771774
bState Key Laboratory for Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: zhanwu@hnu.edu.cn; Fax: +86-731-8822782; Tel: +86-731-8822577
First published on 14th December 2015
Abstract
A novel fluorescence assay strategy has been developed for quantitatively detecting small molecule–protein interactions on the basis of terminal protection. It is well-known that β-indole acetic acid (IAA) is a critical plant hormone molecule regulating plant growth. This terminal protection strategy was demonstrated using IAA and its binding protein anti-IAA antibody as a model case. The IAA-labeled strand of the double-stranded DNA (dsDNA) is protected from degradation by T7 exonuclease when the small molecule moiety is bound to its antibody. By using a competitive assay format, this developed method shows that the fluorescence peaks are dynamically correlated to the concentrations of IAA ranging from 1 to 2000 nM with a detection limit of 0.4 nM. Our assay strategy is able to offer high selectivity, excellent reproducibility, cost-effective, and simplified operations.
Introduction
Detection and quantification of small molecule–protein interactions is of tremendous importance in molecular genetics, clinical diagnostics, and therapeutics.1–3 To date, a variety of methods exist to unravel small molecules and their protein interactions have been developed, including fluorescence resonance energy transfer (FRET),4 affinity chromatography,5 kinetic capillary electrophoresis,6 fluorescence polarization,7 protein-fragment complementation assay,8 and enzyme-linked immunosorbent assay (ELISA).9However, many of the existing methods require substantially improved throughput and decreased time, cost, and material consumption (Table S1 in ESI†). Thus, a universal protocol for cost-effective, highly sensitive, simplified operation and selective detection of small molecules remains a major opportunity and challenge.
It is well-known that IAA is a critical plant hormone molecule regulating the plant growth. Although several assay strategies have been applied in IAA detection,10 such as electrochemistry and fluorescence resonance energy transfer,11 no fluorescence amplification assay strategy using small molecule–protein interaction via terminal protection has been reported as far as we know.12 Considering the poor sensitivities of current analytical techniques, an effective and sensitive analytical protocol for the detection of IAA is especially important to the agriculture and other related fields.
Jiang group has reported on a terminal protection strategy that is conducted by the specific binding of small molecule-labeled DNA and target protein.13 Oligonucleotide probes tethered with specific target molecules provides with the versatile capacity to specifically bind to the target protein. The double-stranded DNA hybrid offers the capacity of signal transduction and amplification.14
This assay is based on our reasoning that the small molecule-linked strand of the double-stranded DNA (dsDNA) is protected from the degradation by T7 exonuclease when the small molecule moiety is bound to its protein target.15 This terminal protection strategy was demonstrated using β-indole acetic acid (IAA) and its binding protein anti-IAA antibody as a model case. T7 exonuclease has been reported to degrade the double-stranded DNA (dsDNA) in the 5′ to 3′ direction.16 Probe 2 with a 5-carboxy fluorescein (FAM) tag at the 5′ end and a BHQ-1 quencher in adjacent nucleotides is designed to perfectly complement the small molecule-linked DNA (probe 1).
The interaction of small molecule-linked DNA (probe 1) with its protein may dramatically increases steric hindrance, which protects the FAM-labeled probe (probe 2)/probe 1 hybrid from T7 exonuclease-catalyzed digestion. This strategy is essentially a competitive assay format between the labeled and free small molecules, which compete for binding the antibody protein.
In the present of free IAA, the free IAA molecules are bound to the anti-IAA antibodies by preincubating in the target sample solution. Thus, the IAA-labeled DNA (probe 1) cannot interact with anti-IAA antibody. Without the terminal protection of the small molecule–protein interactions, T7 exonuclease cyclically cleaves the fluorescence-quenched DNA probe successively into mononucleotides from the 5′ terminal, thereby activating an obviously strong fluorescence signal. Namely, the displaced labeled-probe become available for binding another FAM-labeled probe (probe 2) and triggering new amplification cycles.
Because cyclical cleavage of the input the FAM-labeled probe affords efficient amplification of the fluorescence signal, this strategy is able to ensure substantial signal amplification and a low background current. By using a competitive assay format between the labeled IAA and the free IAA, this developed method shows that the fluorescence emission peaks are dynamically correlated to the concentrations of IAA range from 1 to 2000 nM with a detection limit of 0.4 nM. Such a developed assay technology offers outstanding advantages over existing techniques such as the surface-based methods. Thus, this assay strategy may offer a simple, highly selective, excellent reproducibility, cost-effective, and simplified detection platform for identification and quantification of small molecule–protein interactions.
Experimental
Chemicals and materials
The small molecule-linked DNA (probe 1) oligonucleotides used in this work were synthesized by Takara Biotechnology Co., Ltd. (Dalian, China). The FAM-labeled probe (probe 2) oligonucleotides were synthesized by Sangon Biological Engineering Technology & Service Co., Ltd. (Shanghai, China). Their sequences are listed in Table S2 in ESI.† β-Indole acetic acid (IAA), anti-IAA antibody (mouse monoclonal), abscisic acid (ABA), gibberellin (GA), cytokinin (CTK), 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide (Sulfo-NHS) were all purchased from Sigma-Aldrich (U.S.A.). 5× TBE buffer (225 mM Tris–boric acid, 50 mM EDTA, pH 8.0) was bought from Shanghai Sangon Biotechnology Co. Ltd. (Shanghai, China). Bovine serum albumin (BSA) was purchased from Dingguo Biotech. Co. (Beijing, China). GeneRuler Ultra Low Range DNA Ladder was obtained from Thermo Fisher Scientific Inc. (Waltham, MA, U.S.A.). All other reagents were of analytical grade and were used without further purification. Ultrapure water obtained from a Millipore Milli-Q water purification system (resistance > 18.2 MΩ cm−1) was used throughout the experiments.Labeling of IAA to NH2-modified oligonucleotides
Dissolve the IAA to be activated in 1 mL of 0.01 M PBS, 0.5 M NaCl, pH 6.0 (reaction buffer), at a concentration of 1 mg mL−1. Equilibrate EDC and NHS to room temperature before opening bottles.17 Add 1 mg EDC and 2.5 mg of Sulfo-NHS to 1 mL of IAA solution quickly and react for 10–15 minutes at room temperature. After addition of 2-mercaptoethanol to the reaction solution to quench the EDC reaction, the activated IAA solution could be added directly into the DNA solution for conjugation. Allow the solution to react for 2 hours at room temperature.The conjugate was dialyzed against 10 mM PBS (1000 mL) for 24 h to remove excess by using a membrane with molecular weight cutoff of 1000 Da. The dialysis was performed for 3 days with shielding from light and changes with fresh buffer every 4–5 h.
Real-time fluorescence analysis
Real-time fluorescence analysis was performed in a 30 μL mixture containing 1× T7 exonuclease buffer (20 mM Tris–Ac, 10 mM Mg(Ac)2, 50 mM KAc, pH 7.9), 1 U μL−1 T7 exonuclease, probe 1 (100 nM), probe 2 (500 nM), anti-IAA antibody (100 μg mL−1) or BSA (100 μg mL−1). Time-dependent fluorescence responses were observed under different conditions. (a) Unlabeled DNA probe/probe 2/anti-IAA/T7 exonuclease. (b) Unlabeled DNA probe/probe 2/anti-IAA (c) IAA-labeled DNA probe/probe 2/T7 exonuclease. (d) IAA-labeled DNA probe/anti-IAA/probe 2/T7 exonuclease. (e) IAA-labeled DNA probe/BSA/probe 2/T7 exonuclease. (f) IAA-labeled DNA probe/IAA/anti-IAA/probe 2/T7 exonuclease. The reaction was performed at 25 °C for 120 min on a C1000 Thermal Cycler (Bio-Rad, Hercules, CA, USA) with a CFX96 in suit detection system. The real-time fluorescence intensity was monitored in 30 s intervals using the FAM/SYBR green channel. All samples were measured in triplicate.Gel electrophoresis analysis of terminal protection
To verify terminal protection of IAA-labeled DNA probe 1 and 2 against T7 exonuclease, DNA samples were prepared by adding to 30 μL of 1× T7 exonuclease buffer solution containing 20 mM Tris–Ac, 10 mM Mg(Ac)2, 50 mM KAc (pH 7.9), 2 μM probe 1, probe 2 and anti-IAA antibody. Each sample was incubated at 37 °C for 45 min in the presence or absence of target proteins to allow complete interaction between the protein and the DNA hybrid duplex. The T7 exonuclease digestion was performed at 25 °C for 120 min. Lane M, DNA marker; lane 1, 2 μM unlabeled DNA probe 1 and 5 μM probe 2; lane 2, 2 μM IAA-labeled DNA probe 1 and 5 μM probe 2; lane 3, 2 μM unlabeled DNA probe 1 and 5 μM probe 2 digested by 1 U μL−1 T7 exonuclease; lane 4, 2 μM IAA-labeled DNA probe 1 and 5 μM probe 2 plus anti-IAA antibody with 1 U μL−1 T7 exonuclease. Gel electrophoresis analysis was performed using 5% (w/w) agarose gels containing 0.5 g mL−1 ethidium bromide (EB) in 0.5× TBE buffer. The gel electrophoresis was then performed at a constant potential of 101 V for 75 min with a load of 10 μL of sample in each lane at room temperature. After electrophoresis, the gel was visualized via a Tocan 240 gel imaging system (Shanghai Tocan Biotechnology).Fluorescence assay of small molecule–protein interactions
IAA samples (final concentrations of IAA ranging from 0 to 2000 nM) were added in the a 30 μL reaction mixture containing 1× T7 exonuclease buffer (20 mM Tris–Ac, 10 mM Mg(Ac)2, 50 mM KAc, pH 7.9), probe 1 (100 nM), probe 2 (500 nM), antibody (100 μg mL−1). The reaction was incubated at 37 °C for 45 min to allow complete interaction between IAA and antibody. The T7 exonuclease (1 U μL−1) was subsequently added into the mixture at 25 °C for 2 h before fluorescence assay. The selectivity investigations against non-target plant hormone molecules including abscisic acid (ABA), gibberellin (GA), cytokinin (CTK) were also tested in this assay strategy. The fluorescence measurements were performed on a F-7000 fluorescence spectrometer (Hitachi, Japan). The slit width for both excitation and emission was set at 5 nm. The excitation wavelength was 494 nm and scanning the emission from 505 to 600 nm at room temperature.Results and discussion
This terminal protection strategy was demonstrated using β-indole acetic acid (IAA) and its binding protein anti-IAA antibody as a model case. This working principle is illustrated in Scheme 1. It was reported that the double-stranded DNA (dsDNA) could be degraded by T7 exonuclease in the 5′ to 3′ direction. Probe 2 labeled with a FAM tag at the 5′ end and a BHQ quencher in adjacent nucleotides is designed to perfectly complement the small molecule-linked DNA (probe 1). The small molecule-linked DNA was obtained by conjugating probe 1 with IAA via NH2-modified T nucleotide.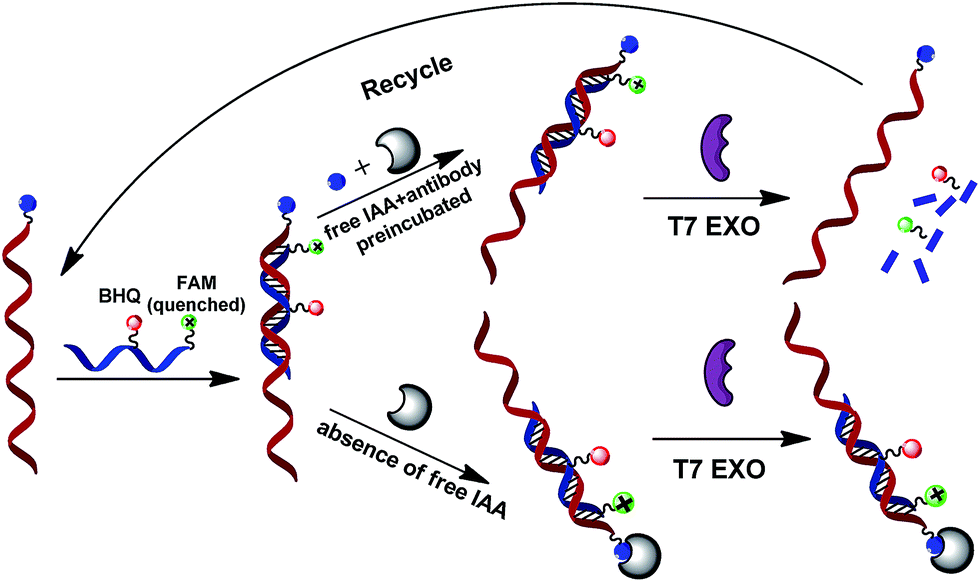 | ||
| Scheme 1 Terminal protection based fluorescence detection of small molecule–protein interactions. | ||
The principle mechanism of terminal protection assay is that binding a protein to small molecule-labeled DNA will may dramatically increases steric hindrance around the binding site, and thus prevent the FAM-labeled probe (probe 2)/probe 1 hybrid from T7 exonuclease-catalyzed digestion. As for the reaction mixture containing target IAA molecules, the free IAA molecules are bound to the anti-IAA antibodies by preincubating in the target sample solution. Thus, the IAA-labeled DNA (probe 1) cannot interact with anti-IAA antibody. Without the terminal protection of the small molecule–protein interactions, T7 exonuclease cyclically cleaves the fluorescence-quenched DNA probe successively into mononucleotides from the 5′ terminal, thereby a significantly strong fluorescence signal was obtained. Namely, the displaced labeled-probe was released to bind another FAM-labeled probe (probe 2) and rendering a new amplification cycle. Due to cyclical cleavage of the input FAM-labeled probe, this efficient fluorescence signal amplification strategy may have a great potential for the detection of small molecule–protein interactions.
It was verified that the terminal protection mechanism could work with T7 exonuclease. To verify terminal protection of IAA-labeled DNA probe 1 and 2 against T7 exonuclease, we design a gel electrophoresis assay strategy. T7 exonuclease has been reported to degrade the double-stranded DNA (dsDNA) in the 5′ to 3′ direction. The interaction of small molecule-linked DNA (probe 1) with its protein may effectively increases steric hindrance, which protects the FAM-labeled probe (probe 2)/probe 1 hybrid from the digestion of T7 exonuclease.
As shown in Fig. 1, the DNA double-strand hybrid duplex generated by the hybridization of unlabeled DNA probe 1 with the probe 2 (lane 1), the double-stranded DNA duplex generated by the hybridization of IAA-labeled DNA probe 1 with the probe 2 (lane 2) exhibited two bright bands with the same mobility.
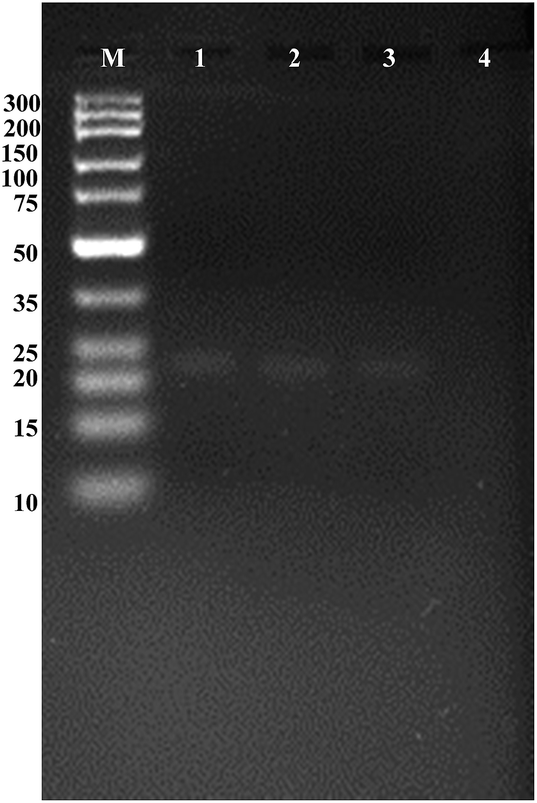 | ||
| Fig. 1 Agarose gel electrophoresis image for terminal protection against T7 exonuclease. Lane M, DNA marker; lane 1, 2 μM unlabeled DNA probe 1 and 5 μM probe 2; lane 2, 2 μM IAA-labeled DNA probe 1 and 5 μM probe 2; lane 3, 2 μM IAA-labeled DNA probe 1 and 5 μM probe 2 plus anti-IAA antibody with 1 U μL−1 T7 exonuclease; lane 4, 2 μM unlabeled DNA probe 1 and 5 μM probe 2 digested by 1 U μL−1 T7 exonuclease. | ||
After adding T7 exonuclease into the reaction mixture above, it was observed that almost no bright band (lane 4) compared with the bright band in lane 3. Because of the degraded DNA mononucleotides would migrate out of the gel and only double-strand DNA oligonucleotides could be stained selectively using ethidium bromide, we could infer that probe 2 was digested by T7 exonuclease into mononucleotides without the terminal protection of IAA molecule–antibody interaction. Thus, the appearance of visible bands in the gel could directly verify the terminal protection of anti-IAA antibody and the labeled IAA could inhibit the action of T7 exonuclease. Considering the existing methods require substantially improved throughput and decreased time, it is of tremendous importance to develop an assay strategy for small molecule detection based on signal transduction and amplification protocol. On the basis of the terminal protection, this sensitive and selective fluorescence assay strategy may hold considerable potential in small molecule–protein interaction investigations.
As shown in Fig. 2, the terminal protection of IAA-labeled DNA probe 1 and 2 against T7 exonuclease was further studied using fluorescence analysis. Each sample was incubated with T7 exonuclease at 25 °C for 100 min before the fluorescence measurement. Without the addition of the anti-IAA antibody, the digested probe 2 separates the fluorophore from the quencher, activates the fluorescence signal. The end-point fluorescence readout showed a dramatically higher fluorescent intensity, compared with the presence of anti-IAA antibody. These datas demonstrated that binding of anti-IAA antibody to the IAA label could prevent the IAA-labeled DNA probe 1/probe 2 hybrid from cleaving.
 | ||
| Fig. 2 Fluorescence intensity in absence (red) and the presence (black) of the anti-IAA antibody. The excitation wavelength was 494 nm and the emission wavelength was from 505 nm to 600 nm. | ||
As shown in Fig. 3, we could clearly see the time-dependent fluorescence responses under different conditions. Each sample was incubated at 37 °C for 45 min before the real-time fluorescence measurement. From Fig. 3A, in the mixture containing only 100 nM unlabeled DNA probe, 500 nM probe 2, 100 μg mL−1 anti-IAA, a very weak fluorescence signal (∼201), was observed after an incubation of 2 h at 25 °C, and the fluorescence readout did not show appreciable time-dependent changes. When adding T7 exonuclease into the systems containing 100 nM unlabeled DNA probe, 500 nM probe 2, 100 μg mL−1 anti-IAA and 1 U μL−1 T7 exonuclease, the fluorescence was remarkably enhanced, and confirming that probe 2 was digested by T7 exonuclease. From Fig. 3B, in the presence of anti-IAA antibody, the mixture showed a much slower rate of fluorescence activation. After an incubation at 25 °C for 2 h, the end-point fluorescence readout showed only a ∼3-fold enhancement, compared with that for the noncleaved probe 2 (line b). We could observed that, in the absence of IAA, this system gave a high signal-to-background ratio compared with the fluorescence intensity for the addition of anti-IAA antibody. This immediate evidence confirmed that the binding of anti-IAA antibody to the IAA label could prevent the IAA-labeled DNA probe 1/probe 2 hybrid from cleaving.
 | ||
| Fig. 3 (A) unlabeled DNA probe/probe 2/anti-IAA/T7 exonuclease (line a); unlabeled DNA probe/probe 2/anti-IAA (line b). (B) IAA-labeled DNA probe/probe 2/T7 exonuclease (line c); IAA-labeled DNA probe/anti-IAA/probe 2/T7 exonuclease (line d). (C) IAA-labeled DNA probe/BSA/probe 2/T7 exonuclease (line e); IAA-labeled DNA probe/IAA/anti-IAA/probe 2/T7 exonuclease (line f). | ||
Furthermore, as shown in Fig. 3C, we replaced the anti-IAA by another protein BSA in the system to test terminal protection of the labeled-IAA and the anti-IAA antibody. We then performed further experiment under the condition of excessive free IAA, it was observed that, the system gave almost the same fluorescence intensity for the mixture of IAA-labeled DNA probe 1, BSA, probe 2 and T7 exonuclease. These results indicated that inhibition of T7 exonuclease digestion activity of the system specifically arises from the binding of anti-IAA antibody to the IAA label.
Fig. 4A shows typical the fluorescence peaks of terminal protection assay in response to IAA of varying concentrations. The fluorescence responses of IAA-labeled biosensor in competitive assay of the free IAA were observed, and the fluorescence peaks were dynamically increased with increasing concentrations of IAA within the range of 1 to 2000 nM.
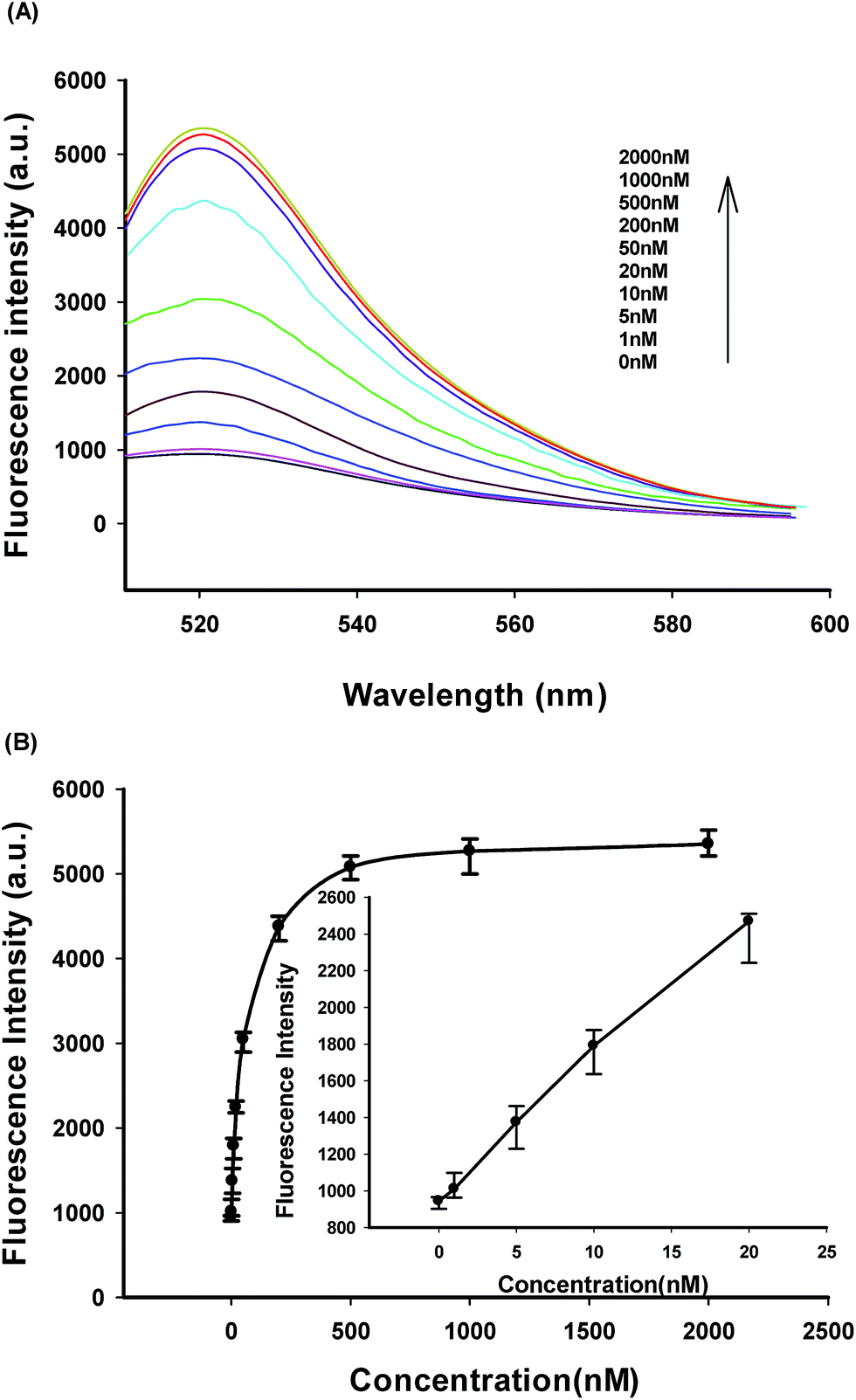 | ||
| Fig. 4 (A) Fluorescence responses of IAA-labeled biosensor in competitive assay of the free IAA. (B) Fluorescence intensity at 520 nm versus the concentration of the free IAA. | ||
As shown in Fig. 4B, the fluorescence peak intensity versus IAA concentrations showed a linear correlation ranging from 0 to 20 nM. The detection limit was estimated to be 0.4 nM according to the 3σ rule. Relative standard deviations (RSDs) of fluorescence intensity at 520 nm were 3.09%, 3.35%, 2.77%, and 2.85% in four repetitive assays of 20, 200, 500, and 2000 nM IAA. Also, the developed strategy exhibited a very desirable reproducibility, high signal-to-background ratio and a wide dynamic response range based on its fluorescence amplification detection assay and competitive assay format for the detection of small molecule–protein interactions.
Specificity evaluation of terminal protection assay strategy for IAA detection was shown in Fig. 5. It was observed that the fluorescence spectrometer yielded very weak fluorescence peaks in response to these plant hormone small molecules such as GA, CTK, ABA, which contrasted clearly with the strong fluorescence response to IAA.
 | ||
| Fig. 5 The selectivity investigations against non-target molecules: blank, ABA, GA, CTK. | ||
In the specificity evaluation using the assay system with different plant hormone small molecules, we obtained very small fluorescence peaks, implying that fluorescence assay strategy was excellent selective.
Conclusion
This study relies on the interaction between anti-IAA antibody and the IAA label, which would inhibit the effective digestion of T7 exonuclease, resulting in low fluorescence intensity. On the basis of the terminal protection mechanism, a novel fluorescence assay strategy was expected to provide a highly sensitive, specific, and efficient platform for quantitatively detecting the small molecule–protein interactions via the efficient signal amplification from the cyclic cleavage operation. This strategy was confirmed that exhibits a high signal-to-background ratio, easy operations, cost-effective, desirable sensitivity and selectivity compared with the previously reported assay strategies. In view of these advantages, the proposed assay may serve as a versatile tool in multiplex investigations.Acknowledgements
This work was supported by the General Armament Department of Weapons and Equipment, Pre-research Fund (9140A26030214JB11418).Notes and references
- P. C. Lin, M. C. Tseng, A. K. Su, Y. J. Chen and C. C. Lin, Anal. Chem., 2007, 79, 3401–3408 CrossRef CAS PubMed.
- X. Duan, Y. Li, N. K. Rajan, D. A. Routenberg, Y. Modis and M. A. Reed, Nat. Nanotechnol., 2012, 7, 401–407 CrossRef CAS PubMed.
- E. N. Imyanitov, Hum. Genet., 2009, 125, 239–246 CrossRef PubMed.
- M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki and K. T. Douglas, J. Am. Chem. Soc., 2008, 130, 5720–5725 CrossRef CAS PubMed.
- J. Taunton, C. A. Hassig and S. L. Schreiber, Science, 1996, 272, 408–411 CAS.
- K. C. Electrophoresis, J. Am. Chem. Soc., 2005, 127, 17104–17110 CrossRef PubMed.
- D. Axelrod, Biophys. J., 1979, 26, 557 CrossRef CAS PubMed.
- S. W. Michnick, P. H. Ear, E. N. Manderson, I. Remy and E. Stefan, Nat. Rev. Drug Discovery, 2007, 6, 569–582 CrossRef CAS PubMed.
- P. W. White, S. Titolo, K. Brault, L. Thauvette, A. Pelletier, E. Welchner, L. Bourgon, L. Doyon, W. W. Ogilvie and C. Yoakim, J. Biol. Chem., 2003, 278, 26765–26772 CrossRef CAS PubMed.
- V. C. Pence and J. L. Caruso, Phytochemistry, 1987, 26, 1251–1255 CrossRef CAS.
- J. Li, Z.-Y. Wu, L.-T. Xiao, G.-M. Zeng, G.-H. Huang, G.-L. Shen and R.-Q. Yu, Anal. Sci., 2002, 18, 403–407 CrossRef CAS PubMed.
- E. Weiler, P. Jourdan and W. Conrad, Planta, 1981, 153, 561–571 CrossRef CAS PubMed.
- Z. Wu, Z. Zhen, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 12325–12332 CrossRef CAS PubMed.
- D. Guiffant, D. Tribouillard, F. Gug, H. Galons, L. Meijer, M. Blondel and S. Bach, Biotechnol. J., 2007, 2, 68–75 CrossRef CAS PubMed.
- Z. Wu, H. Wang, M. Guo, L.-J. Tang, R.-Q. Yu and J.-H. Jiang, Anal. Chem., 2011, 83, 3104–3111 CrossRef CAS PubMed.
- K. Shinozaki and O. Tuneko, Nucleic Acids Res., 1978, 5, 4245–4262 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic press, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DNA sequences and additional figures. See DOI: 10.1039/c5ra20240j |
| This journal is © The Royal Society of Chemistry 2015 |