
C-8 Mannich base derivatives of baicalein display improved glucuronidation stability: exploring the mechanism by experimentation and theoretical calculations†
Abstract
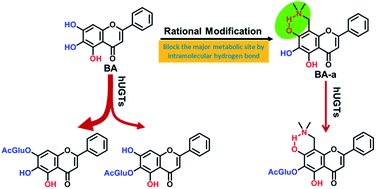
Baicalein (BA), a natural flavonoid compound, possesses many desirable pharmacological activities. However, poor solubility and extensive metabolism by human UDP-glucuronosyltransferases (UGTs) strongly restrict the clinical applications of BA. We previously reported that two C-8 Mannich base derivatives of BA (BA-a and BA-j) displayed enhanced solubility and anti-cyclin dependent kinase 1 activity, yet the metabolic stabilities of these compounds remained unknown. This study aimed to evaluate the in vitro glucuronidation stability of these BA derivatives and to explore the key factors affecting the UGT-mediated biotransformation. The results showed that the glucuronidation stabilities of these BA derivatives were much higher than BA. BA-a exhibited 12-fold and BA-j exhibited 5-fold improved stability in human liver S9, while in human intestine S9, BA-a and BA-j exhibited 42-fold and 33-fold improved stability, respectively. Further investigations found that the major glucuronidation site(s) were changed from 7-OH and 6-OH in BA to 6-OH in the BA derivatives. Also, both the involved enzymes and their catalytic efficacy in 6-O-glucuronidation of BA derivatives were much lower than that of BA. The formation of an intramolecular hydrogen bond between the C-8 Mannich base substituents and C-7 phenolic groups played a predominant role in these glucuronidation changes. The calculated bond dissociation energy (BDE) of each phenolic group in BA and its derivatives agreed well with their glucuronidation activities. All these findings bring new insights into the structure–glucuronidation relationship and provide a practical strategy for the structural modification to improve the glucuronidation stability of drug candidates, especially for those phenolic compounds.
 Please wait while we load your content...
Please wait while we load your content...

