
Cu2−xS/graphene oxide nanocomposites for efficient photocatalysis driven by real sunlight
Abstract
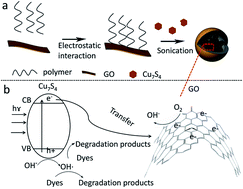
We present Cu2−xS/graphene oxide nanocomposites, which exhibit excellent photocatalytic properties toward photodegradation of organic compounds by real sunlight due to the broadband absorption of Cu2−xS and good electron trapping and shuttling ability of graphene.
 Please wait while we load your content...
Please wait while we load your content...

