
Nanostructured oxytyramine catalyst for the facile one-pot synthesis of cyclohexanecarbonitrile derivatives
Abstract
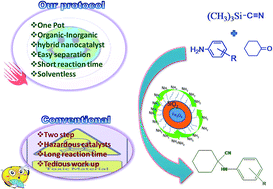
The magnetic, recyclable heterogeneous organocatalyst OT@Si@SPIONs has been developed in this report, with the aim of synthesizing cyclohexanecarbonitriles. The prepared nanocatalyst was fully characterized by various techniques and its catalytic activity has been tested in a one pot reaction which involves the in situ formation of imines from cyclohexanones and amines, which further undergo nucleophilic addition with TMSCN. Moreover, although this Strecker reaction is centuries old, its applicability to ketones is still a less explored subject, and our investigation provides an insight into the efficient synthesis of cyclohexanecarbonitrile derivatives using a ketone i.e. cyclohexanone. When compared with other catalytic systems reported in the literature, ours showed superior catalytic activity at a remarkably low catalyst loading i.e. 5 mg.
 Please wait while we load your content...
Please wait while we load your content...

