
Synthesis and antibacterial activity of ricinoleic acid glycosides†
Ramakrishna Kuppalaa,
Mugunthan Govindarajana,
Rushikesh Tambatb,
Neeraj Patelc,
Hemraj Nandanwar*b,
Kamlesh K. Bhutanic and
K. P. Ravindranathan Kartha*a
aDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, S.A.S. Nagar, Punjab-160062, India. E-mail: rkartha@niper.ac.in; Fax: +91-172-2214692; Tel: +91-172-2292028
bBioactive Screening Laboratory, CSIR-Institute of Microbial Technology, Sector-39A, Chandigarh-160036, India. E-mail: hemraj@imtech.res.in; Fax: +91-172-2690585; Tel: +91-172-6665338
cDepartment of Natural Products, National Institute of Pharmaceutical Education and Research, S.A.S. Nagar, Punjab-160062, India
First published on 23rd December 2015
Abstract
The antibacterial properties of twenty-eight novel ricinoleic acid glycosides synthesized by Koenigs–Knorr glycosylation are reported. Seven of them were found to show promising wide spectrum antibacterial activity against Gram positive bacteria of which two compounds, the mannopyranosyl- and the arabinofuranosyl derivatives, were proven effective against various non-clinical/clinical/NorA-overexpressed/resistant strains of Staphylococcus aureus as well as other Gram +ve bacteria such as Bacillus subtilis ATCC 6051 and Micrococcus luteus MTCC 2470. It was found that both the presence of the sugar and its structure are necessary and important for the compounds to be bioactive. The methyl ester protection of the carboxylic acid moiety of the ricinoleic acid unit was also found to be important for imparting good bioactivity to the molecule. Based on the membrane permeability and cell disintegration studies, these compounds are found to increase the bacterial cell membrane permeability, subsequently causing cell death.
Introduction
Traditionally, man has relied heavily on natural products for alleviating problems associated with diseases and infections caused by various microbes. It is therefore not surprising that many of the antibiotics currently in use are natural products or natural product-based/inspired. Although, following a surge in the development and marketing of antibiotics that was witnessed during the first three quarters of the last century, it was suggested during the fourth quarter of the century that there were perhaps no unmet needs in the antibiotic therapeutics,1 however, a renewed search for new antibiotic molecules returned by the turn of the century, largely due to the increasing emergence of instances of drug resistance.2–4 In fact antibiotic-resistant infections have now become a major health concern globally. Thus, the incidents of drug resistant cases of methicillin resistant Staphylococcus aureus (MRSA),5,6 as well as multidrug drug resistant (MDR) and extensively drug resistant (XDR) tuberculosis (TB)7 are being reported worldwide and hence the demand for identifying drug candidates with novel modes of action has been growing.7,8 Many of the drugs used for the treatment of bacteria including mycobacterial infection are cell wall biosynthesis inhibitors.9 The bacterial cell wall possesses a complex architecture made up of, besides others, glycans constituted of sugars such as N-acetylglucosamine, mannose, galactopyranose (Galp), galactofuranose (Galf), arabinofuranose, ribose and rhamnose,10 and use different biosynthetic pathways, many of which are validated drug targets, for the construction of these glycan structures.The antimicrobial activity of unsaturated fatty acids such as oleic/linoleic/linolenic/arachidonic acid, etc., as for example evident from their use as effective antimicrobial food additives, is well known.11,12 Likewise, ricinoleic acid (12-hydroxy-9-cis-octadecenoic acid, 1, Fig. 1), found in castor oil is also well-known for its medicinal values and commercial importance. The first report of ricinoleic acid derivative, for example, sodium ricinoleate (2a, Fig. 1) was as a surface tension depressant on the growth of bacterial culture media.13,14 Its ability to act as an antibacterial agent against organisms such as Corynebacterium diphtheriae, Gonococci, etc. has also been demonstrated.15–17 Potassium and zinc ricinoleates (2b and 4, Fig. 1) were also shown to be antibacterial in nature.15 Presently ricinoleic acid and its various salts/derivatives have reportedly been used in more than 750 commercial products18 in which they also fulfill their role as emulsion stabilizers, cosmetics, skin-conditioning agents, etc. Also, mono- and diglycerides of ricinoleic acid have been reported as potential antimycobacterial agents.19 D'Oca et al. reported different fatty acid amides and their structure activity relationship in the context of anti-TB activity and ricinoleic acid amide (5, Fig. 1) was among those exhibited promising anti-TB activity against M. tb H37Rv.20 More recently, during the progress of this work, certain Schiff base analogues of methyl ricinoleate were shown to be good antimicrobials against S. aureus.21
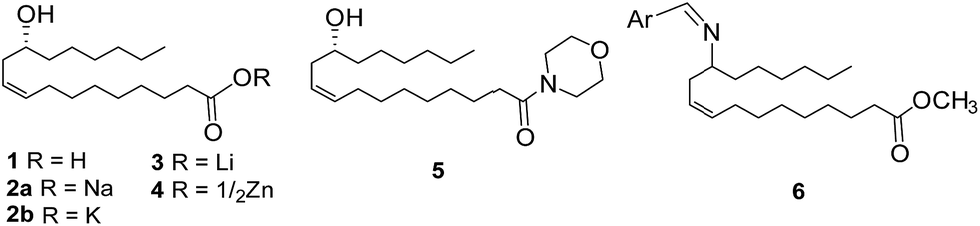 | ||
| Fig. 1 Ricinoleic acid-derived compounds known in the literature as anti-bacterial and antimycobacterial agents. | ||
The present work was aimed at synthesizing a library of various glycosides of ricinoleic acid with a view to evaluating their potential as antibacterial agents. D-Glucose, D-galactopyranose, D-mannose, N-acetyl-D-glucosamine, L-rhamnose, D-arabinose, D-galactofuranose and D-lactose were chosen as the sugar substrates to be linked glycosidically to methyl ricinoleate.
Results and discussion
Methyl ricinoleate (7), prepared by reacting 1 with MeOH in the presence of anhydrous HCl,22 was chosen as the acceptor alcohol for glycosylation with various acetohalosugars (8) essentially under the traditional Ag2CO3–AgClO4-mediated Koenigs–Knorr conditions to afford the desired 1,2-trans-linked glycosides (9) in good yields (Scheme 1). The structure of these compounds were confirmed by NMR spectroscopy in which the J1,2 value of 7.9–8.0 Hz was typical, for example, of the 1,2-trans-linked H-1 and H-2 for the galacto- and gluco-configured pyranosides. Likewise, the manno-, arabino-, rhamno- and the galactofurano-compounds also gave the J1,2 values characteristic of them (1.5–1.7 Hz, see ESI† for details). It was observed that among the three hexose-derived acetobromosugars (13–15) the comparatively less reactive gluco-compound (14), not surprisingly though, did also yield some orthoester (9a) as a by-product (see ESI† for details). The acyl group deprotection of these glycoside-derivatives was done by trans-esterification using methanolic NaOMe to afford 10. Compound 10 upon aqueous alkaline hydrolysis yielded the desired glycosylated ricinoleic acids 11 in the form of their metal salts corresponding to the alkali employed for the hydrolysis.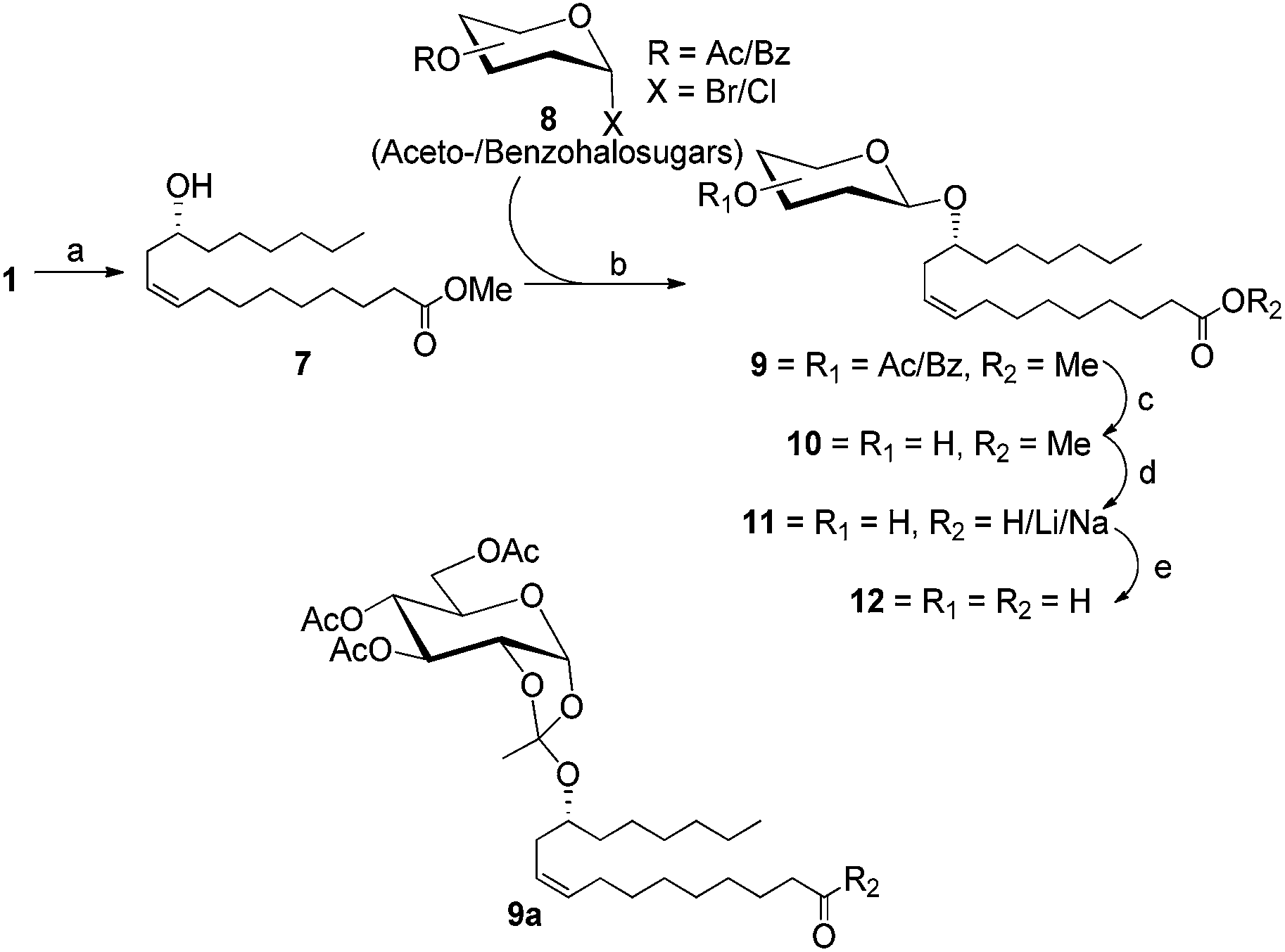 | ||
| Scheme 1 Synthesis of ricinoleic acid glycosides in their partially/fully deprotected form and/as their sodium/lithium salts. Reagents and conditions: (i) HCl/MeOH, rt, 4 h, 90%; (ii) Ag2CO3, AgClO4, powdered molecular sieves (4 Å), CH2Cl2 rt, 10–12 h, 53–68%; (iii) NaOMe/MeOH, 20–40 min, rt, 90–94%; (iv) LiOH/aq THF, 2–4 h, rt, 80–87%; (v) Amberlite IR 120 (H+), rt, 80–88%. | ||
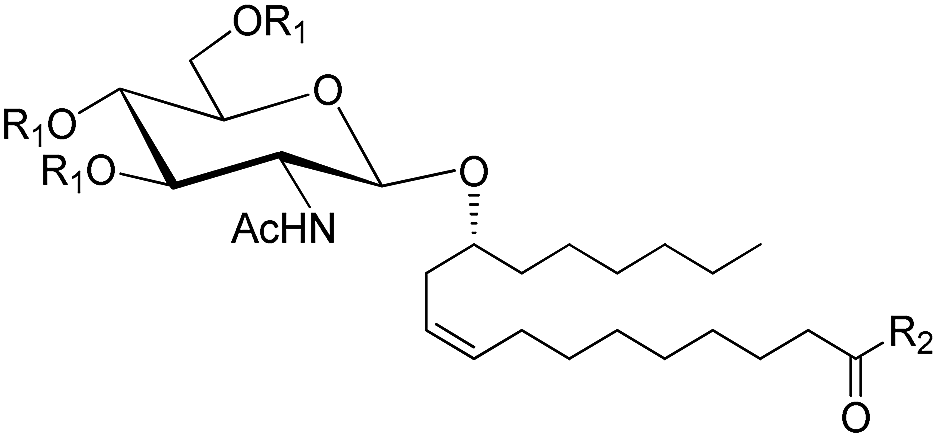
When the free acid was required, the salts obtained were deionized using Amberlite IR 120 H+ resin (Scheme 1). The compounds synthesized (21–48) are shown in Table 1.
| Acetohalosugar used for synthesis | Glycosylated ricinoleates synthesized | Yield (%) | |
|---|---|---|---|
| Structure | Compound number | ||
 |
 |
21, R1 = Ac, R2 = OCH3 | 68 |
| 22, R1 = H, R2 = OCH3 | 92 | ||
| 23, R1 = H, R2 = OH | 87 | ||
 |
 |
24, R1 = Ac, R2 = OCH3 (ref. 25) | 58 |
| 25, R1 = H, R2 = OCH3 | 90 | ||
| 26, R1 = H, R2 = OH | 81 | ||
| 27, R1 = H, R2 = O−Li+ | 83 | ||
 |
 |
28, R1 = Ac, R2 = OCH3 | 60 |
| 29, R1 = H, R2 = OCH3 | 94 | ||
| 30, R1 = H, R2 = OH | 85 | ||
| 31, R1 = H, R2 = O−Na+ | 80 | ||
 |
 |
32, R1 = Ac, R2 = OCH3 | 52 |
| 33, R1 = H, R2 = OCH3 | 92 | ||
| 34, R1 = H, R2 = OH | 83 | ||
 |
 |
35, R1 = Bz, R2 = OCH3 | 66 |
| 36, R1 = H, R2 = OCH3 | 91 | ||
| 37, R1 = H, R2 = OH | 87 | ||
 |
 |
38, R1 = Bz, R2 = OCH3 | 57 |
| 39, R1 = H, R2 = OCH3 | 92 | ||
| 40, R1 = H, R2 = OH | 79 | ||
 |
 |
41, R1 = Ac, R2 = OCH3 | 60 |
| 42, R1 = H, R2 = OCH3 | 94 | ||
| 43, R1 = H, R2 = OH | 85 | ||
 |
 |
44, R1 = Ac, R2 = OCH3 | 53 |
| 45, R1 = H, R2 = OCH3 | 91 | ||
| 46, R1 = H, R2 = OH | 84 | ||
| 24 (for the structure, see the entry at row 2 above) |  |
47, R1 = Ac, R2 = OCH3 | 80 |
| 48, R1 = H, R2 = OCH3 | 87 | ||

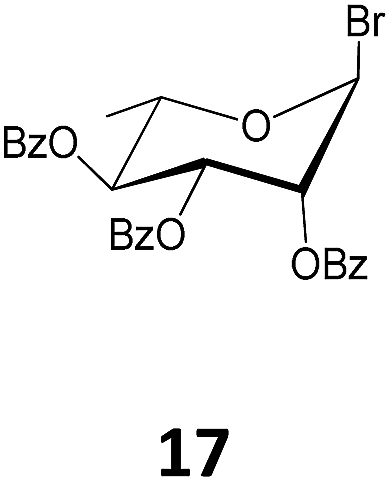
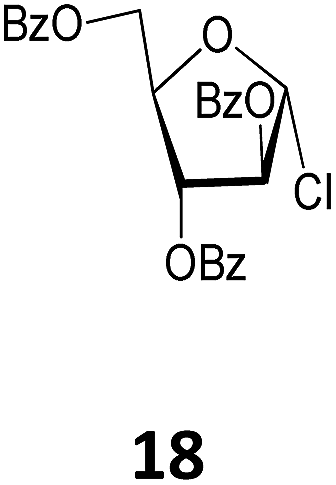
While acetobromosugars (13–15 and 20) were used as starting materials for the synthesis of the hexopyranose-based 21, 24, 28 and 44 (mono- and disaccharide derivatives), the acetochlorosugar donors 16 and 19 were found more convenient for the preparation of the corresponding glucosaminide and the galactofuranoside derivatives 32 and 41 as shown in Table 1. On the other hand for the rhamnopyranosyl and the arabinofuranosyl ricinoleates 35 and 38, the easily accessible benzobromo- and benzochlorosugar donors 17 and 18, respectively, were utilized for the crucial glycosylation reaction.
In view of the presence of the double bond in the compounds described above, choosing 24 as a model compound, the easily obtainable dibromo derivative 47 (and 48 derived therefrom) was also synthesized from 24 (Table 1, last entry). As expected 47 obtained on addition of molecular bromine to the double bond in 24 was in the form of a pair of enantiomers (as also the derived 48) as evident from the NMR spectra of 47. Further, in order to establish the importance (or otherwise) of the sugar moiety in the biological activity to be studied, a set of ether analogues 49 (benzyl ether) and 50 (methyl ether) were also synthesized from methyl ricinoleate 7. It must be pointed out that for the synthesis of the 12-O-benzyl ricinoleate 49 from 7 and BnBr neither the method using Ag2O23 nor the one using Cs2CO3 (ref. 24) was successful. However, the reaction of the two (7 and BnBr) promoted by NaH yielded the desired product 49 in admixture with 49a as an inseparable mixture in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 as determined by the 1H NMR spectroscopy of the chromatographically isolated mixture (see ESI† for details). The mixture was therefore subjected to the conditions of Zemplen transesterification in MeOH whereby the benzyl ester 49a was transformed into the methyl ester analogue 49 thus affording the latter overall neatly.
:0.5 as determined by the 1H NMR spectroscopy of the chromatographically isolated mixture (see ESI† for details). The mixture was therefore subjected to the conditions of Zemplen transesterification in MeOH whereby the benzyl ester 49a was transformed into the methyl ester analogue 49 thus affording the latter overall neatly.
Likewise, the reaction of 7 with MeI (promoted by NaH) was also successful in yielding the methyl ether analogue 50 but, again, along with an inseparable by-product. But unlike 49a, the by-product in the latter case was a mixture of 50 (ref. 26) and the lactone 50a (in a ratio of 1:0.5) as characterized by 1H NMR spectroscopy (see ESI† for details). The separation of the lactone (50a) from the ester (50) could however be very successfully facilitated by methanolysis of the lactone (to yield 7) using anhydrous methanolic hydrogen chloride followed by purification by column chromatography.
Evaluation of antimicrobial activity and observations on the structure activity relationship (SAR)
Once in hand, the compounds described above were evaluated for their antimicrobial activity. In the initial screening they were tested for their activity against three Gram +ve bacteria, viz. Micrococcus luteus (MTCC-2470), Staphylococcus aureus (MTCC-96), and Bacillus subtilis (MTCC-121), three Gram −ve bacteria, viz. E. coli (MTCC-739), Pseudomonas aeruginosa (MTCC-2453), Klebsiella planticola (MTCC-530) and a fungus, Candida albicans (MTCC-3017). The evaluations were done on the basis of the zone of inhibition (results not shown here, see ESI† for details) in the usual manner. The deacylated glycosides of methyl ricinoleate 22, 25, 29, 36, 39, 42, 45, and 48 showed good inhibition of the Gram +ve bacteria evaluated. However, the analogous N-acetyl-glucosamine-linked methyl ricinoleate 33 was proved inactive. Also, the glycosides of methyl ricinoleate showed better inhibition as compared to the simple ether analogues methyl 12-O-benzyl ricinoleate (49) and methyl 12-O-methyl ricinoleate (50). Neither methyl ricinoleate (7) nor ricinoleic acid/its lithium/sodium salt (1/3/2a) were also active against any of the strains tested above (see ESI† for details). This showed the importance of the presence of sugar unit in the structure for the antimicrobial activity. Smith et al. while evaluating the antimicrobial activity of 6-O-substituted fatty alkyl ethers/fatty acyl esters of methyl glucopyranosides similarly had observed the importance of the presence of the sugar residue (as well as its nature) in the molecule for significant antimicrobial activity.27 Again, neither the acyl-protected glycosides of methyl ricinoleate 21, 24, 28, 32, 35, 38 and 41 nor the fully deprotected ricinoleic acid glycosides (as free acids) 23, 26, 30, 37, 40, 43 and 46 were active against either of the Gram +ve or the Gram −ve bacteria tested. However, compounds 37 (the rhamnosylated compound) and 44 (the lactose-linked compound) showed moderate inhibition against Bacillus subtilis (MTCC-121). This revealed the importance of ricinoleic acid present in the form of ester in imparting the inhibitory activity. In the work of Smith et al. referred to above also, they had observed that the 6-O-acyl glucosides were more active than the corresponding ethers.27 The deacylated 9,10-dibromo compound 48 (as racemic mixture) showed better inhibition against M. luteus (MTCC-2470) and S. aureus (MTCC-96) compared to the corresponding acylated compound 47 which was inactive. Moreover, the sodium/lithium salts of the deacylated ricinoleic acid glycosides 27 and 31 were also inactive as were the salts of the acid (2 and 3) themselves. Based on these initial results compounds 22, 25, 29, 36, 37, 39, 42, 44, 45 and 48 were taken up for the determination of their MIC values against different Staphylococcal strains along with some of the standard drugs in use for comparison. Of these glycosides studied 37, the L-rhamnosylated methyl ester, and 44 and 45, the lactosylated methyl esters, showed high MIC values (>1000, results not shown) against the non-clinical S. aureus MTCC-96. This reiterated the fact that not only that the acid has to be in its methyl ester form but also that not all sugars are suitable as substituents on the ricinoleic acid derivatives for them to be sufficiently biologically active. The above experiments thus clearly elucidated the importance of the structure of the sugar moiety present in the molecule. The poorer activity of the free acid 37 is in line with the fact that ester form of the acid is necessary for the compound to be effectively active. Likewise, the bulky disaccharide units in 44 and 45 were also proven to be detrimental for good activity. Based on these results compounds 37, 44 and 45 were excluded from further in vitro screening experiments on the bacterial strains. Instead, for ascertaining the generality of application of these compounds to other Gram +ve bacterial strains, Bacillus subtilis and Micrococcus luteus strains were included in the study. The results obtained for the promising compounds chosen are summarized in Table 2 and clearly demonstrates the effectiveness of these compounds to act against all the Gram +ve bacteria tested.| Entry | Test compounds/antibiotics | MIC (μg mL−1) | |||||
|---|---|---|---|---|---|---|---|
| S. aureus MTCC-96 ∼ ATCC 9144 (non-clinical) | MRSA 831 (clinical isolate) | S. aureus 1199 (wild type clinical isolate) | S. aureus 1199B (NorA overproducing strain) | Bacillus subtilis (MTCC 121 ∼ ATCC 6051) | Micrococcus luteus (MTCC 2470) | ||
| 1 | 22 | 16 | 1000 | 8 | 125 | 16 | 16 |
| 2 | 25 | 8 | 1000 | 8 | 1000 | 2 | 2 |
| 3 | 29 | 2 | 1000 | 2 | 32 | 0.25 | 0.5 |
| 4 | 36 | 4 | 1000 | 250 | 1000 | 0.125 | 0.5 |
| 5 | 39 | 4 | 32 | 8 | 4 | 0.5 | 1 |
| 6 | 42 | 4 | 1000 | 2 | 8 | 1 | 2 |
| 7 | 48 | 32 | 1000 | 62.5 | 125 | 2 | 4 |
| 8 | Erythromycin | 0.78 | 25 | 0.39 | 6.12 | 0.5 | 1 |
| 9 | Teicoplanin | 1.56 | 12.5 | 0.19 | 3.125 | 0.25 | 0.5 |
| 10 | Norfloxacin | 0.39 | 100 | 0.78 | 50 | 0.5 | 2 |
| 11 | Oxacillin | 3.12 | 2000 | 0.48 | 1000 | 0.5 | 1 |
| 12 | Ampicillin | 32 | 250 | 1 | 4 | 4 | 16 |
| 13 | Amoxicillin | 16 | 250 | 0.5 | 2 | 2 | 4 |
| 14 | Linezolid | 1.56 | 3.9 | 1.95 | 7.8 | 1 | 2 |
| 15 | Vancomycin | 1.56 | 3.12 | 0.78 | 6.25 | 0.125 | 1 |
Possibly the glycosides of methyl ricinoleate would have permeabilized the bacterial cell membrane more effectively due to their better surfactant properties11 and thus showing the antibacterial activity. As it has been reported in the past, another possible cause for the antibacterial properties of these compounds could be the inhibition of fatty acid biosynthesis (Fab1) enzyme which is an essential component of the fatty acid synthesis in Gram +ve bacteria.11,28,29 Among the promisingly active compounds, 29 and 39 showed (entries 3 and 5, Table 2) a wide spectrum antibacterial activity against different Staphylococcal species. It is possible that the glycolipids 29 and 39 may be playing some specific role in the bacterial cell wall biosynthesis due to the presence of mannose and arabinose in them. They could also possibly act by inhibiting the transferases (or any other glycan-forming enzymes) involved in the bacterial cell wall biosynthesis. To determine the above mentioned possible mechanism of action of these glycosides of ricinoleic acid a set of experiments with compounds 29, 39 and vancomycin (selectively active against Gram +ve bacteria, as the positive control) was carried out as described below.
The binding of vancomycin to D-Ala-D-Ala prevents cell wall synthesis of the long polymers of N-acetylmuramic acid and N-acetylglucosamine that form the backbone strands of the bacterial cell wall, and it prevents the backbone polymers that do manage to form cross-linking with each other. Since the MIC of vancomycin is more or less matching with 29 and 39, we used these compounds for membrane permeability assay as well as cell membrane disintegration assay (see below). The cell permeability effect of 29 and 39 is comparable with vancomycin, a membrane active antibiotic. The uniform increase in the absorbance of o-nitrophenol suggests that the non-membrane permeable o-nitrophenyl-β-galactoside (ONPG) is slowly permeabilized inside the bacterial cells and subsequently is hydrolyzed by the intracellular β-galactosidase to o-nitrophenol that shows absorbance at 410 nm. As shown in Fig. 2 compounds 29 and 39 induced an increase in the permeability of S. aureus cytoplasmic membrane over a period of time in a manner comparable to that of vancomycin. Moreover, the test compounds as well as vancomycin possessed similar MIC values against S. aureus. This suggests that the cytoplasmic membrane would have been permeabilized by compounds 29 and 39.
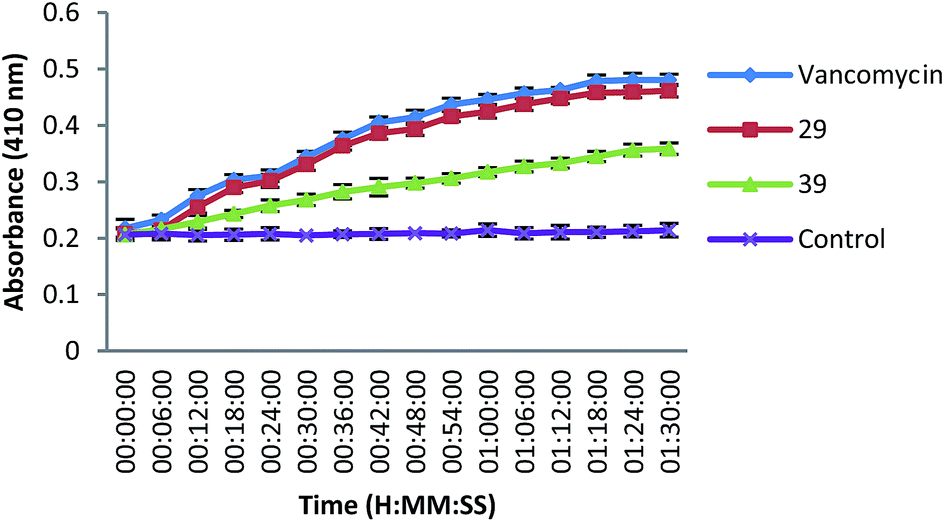 | ||
| Fig. 2 The cytoplasmic membrane permeabilization of S. aureus cells treated with compounds 29 (squares), 39 (triangles) and vancomycin (diamonds). The untreated S. aureus cells (crosses) were taken as control. (Compounds 29 and 39 induced an increase in the permeability of S. aureus. The experiment was carried out two times in triplicate. The results are presented as mean ± SD.) | ||
To verify whether the cells are permeabilized or disintegrated, the propidium iodide (PI) uptake assay was carried out. PI is a viability fluorescent marker that can penetrate impaired cells and intercalate into nucleic acids. Thus, compounds 29-, 39- and vancomycin-induced membrane damage of S. aureus cells, was determined by treating the cells with PI at 37 °C for half an hour using Synergy reader (BioTek, USA). As shown in Fig. 3, in the absence of any antibacterial agent, the untreated control cells of S. aureus showed no PI fluorescence signal. However a significant increase in the PI fluorescence signal was observed in the case of 29, 39 and vancomycin. These results indicate that the membrane integrity of S. aureus cells was affected by the treatment with the test compounds as comparable to that of vancomycin.
 | ||
| Fig. 3 Effect of 29 (squares), 39 (triangles) and vancomycin (diamonds) on membrane integrity of S. aureus cells by propidium iodide uptake assay. The untreated S. aureus cells (crosses) were taken as control. For each sample 106 CFU mL−1 were analyzed. The membrane integrity of S. aureus cells was destroyed by 29 and 39. The experiment was carried out two times in triplicate. The results were presented as mean ± SD. | ||
The cytoplasmic membrane is the main barrier that limits the distribution and entry of antibiotics. In addition to the antimicrobial activities, some serve as an anti-resistance compounds and are able to interact with bacterial membranes, and create ion permeable channels leading to an increased cytoplasmic membrane permeability and hence, bacterial cell death.30 In the case of 29 and 39, they showed permeabilization effect on S. aureus membrane and increased the plasma membrane permeability for entry of ONPG into cells. Moreover, the gradual increase in the fluorescence due to the influx of PI into the cells, indicates that the cytoplasmic membrane could be the most probable target of action of ricinoleic acid glycosides.
Cytotoxicity
The compounds that showed good MIC values in the antibacterial activity evaluation were taken for the cell viability study with a view to assessing their toxicity characteristics. According to FDA castor oil is classified as a safe and effective stimulant laxative18 and it was hoped that attaching a sugar molecule would not result in seriously altering its cytotoxic tolerance. It was therefore not surprising that the synthesized glycosides of methyl ricinoleate 22, 25, 29, 33, 36, 39, 42, 45, and 48 exhibited no significant cytotoxicity when tested up to 500 μg mL−1 (cell viability > 75%) on J774A.1 cells as shown in Fig. 4. However, at higher concentrations of 1000 μg mL−1 compounds 25, 42, 45, and 48 demonstrated cell viability of only 56.2, 68.4, 67.5 and 56.8% respectively. Thus, the results are indicative of good potential for these compounds to be studied in detail further. | ||
| Fig. 4 Cytotoxicity study of biologically active glycosides of methyl ricinoleate (results are expressed as mean ± SD of three independent experiments performed in triplicates). | ||
Experimental
Materials and methods
All reagents and chemicals were purchased from Sigma-Aldrich and were used without further purification. TLC analyses were performed on 0.2 mm Merck pre-coated silica gel 60 F254 aluminium sheets and the spots were visualized under UV lamp and/or by immersion in an ethanolic solution of sulphuric acid (5%, v/v) followed by heating. Final purifications were performed using silica gel 200–400 mesh size. Specific rotations were recorded on a Rudolph Autopol IV Polarimeter at 20 °C. NMR spectra were recorded on a Bruker Avance DPX (400 MHz) spectrometer. 1H NMR and 13C NMR spectra were referenced to the internal standard tetramethylsilane, in the respective deuterated solvents. Coupling constants (J) are reported in Hertz. All assignments were confirmed with the aid of two-dimensional 1H–1H (COSY) and/or 1H–13C (HSQC) experiments using standard pulse programs. Processing of the spectra was performed with MestReNova software. High resolution mass spectra (HRMS) were recorded on a Bruker Maxis spectrometer. Biosafety Cabinet (Clean Air, Chennai, India), CO2 incubator (WTC Binder, Tuttlingen, Germany), Ultracentrifuge (Sigma, St. Louis, MO, USA), autopipettes, ELISA plate reader (Labsystems, Helsinki, Finland) and Neubauer chamber (HBG, Gießen, Germany) were used for the cell culture.Materials and methods for antimicrobial evaluation
000 units mL−1 penicillin and 10 mg mL−1 streptomycin in 0.9% saline, in a CO2 incubator (5% CO2 in air) at 37 °C. The conventional MTT assay was carried out to assess the cell viability of J774A.1 cells using previously reported method.35 All test samples dissolved in DMSO and 0.05% DMSO was used as the control group. The cell viability was calculated in respect to that of control (% of control).General experimental procedures
:hexanes as the eluent to yield the respective glycosides (21/24/28/32/35/38/41/44, respectively) of methyl ricinoleate.Lithium ricinoleate (3). The title compound 3 was prepared from 2 (500 mg, 1.6 mmol), THF (10 mL) and aqueous LiOH solution (1 M, 1.5 mL) by the method (4 h) described above in a yield of 83% (153 mg) as a white fluffy solid. 1H NMR (400 MHz, DMSO) δ 5.43–5.33 (m, 2H, H-9, H-10) 3.47–3.43 (m, 1H, H-12), 2.09–2.06 (m, 2H, 2 × H-11), 1.97 (q, 2H, J = 6.4 Hz, J = 6.5 Hz, 2 × H-8), 1.90 (t, J = 7.4 Hz, 2 × H-2) 1.46–1.23 (m, 20H, 2 × H-7, 2 × H-13 –(CH2)8–), 0.87 (t, J18,17 = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, DMSO) δ 175.65 (C-1), 130.58 (C-9) 126.64 (C-10), 69.84 (C-12), 38.16 (C-2) 36.48 (C-11), 35.23 (C-8), 31.43, 29.45, 29.22, 29.11, 28.93, 28.84, 26.93, 26.30, 22.14, (m, –(CH2)10–), 14.02 (C-18); HR MS m/z calculated for C18H33LiO3 [M + Na]+ = 327.2487, found 327.2480.
Methyl 12-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-ricinoleate (21). The title compound was prepared from 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide (13, 1.00 g, 2.4 mmol) by the general procedure described above. The reaction time was 14 h; and 21 (1.05 g) was obtained in 68% yield as a thick syrup. [α]23D = −1.5° (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.46–5.38 (m, 1H, H-9′), 5.38–5.30 (m, 2H, H-4, H-10′), 5.19 (dd, J2,3 = 10.5 Hz, J2,1 = 7.9 Hz, 1H, H-2), 5.00 (dd, J3,2 = 10.5 Hz, J3,4 = 3.5 Hz, 1H, H-3), 4.50 (d, J1,2 = 7.9 Hz, 1H, H-1), 4.16 (dd, J6a,6b = 11.2 Hz, J6a,5 = 6.8 Hz, 1H, H-6a), 4.09 (dd, J6b,6a = 11.2 Hz, J6b,5 = 6.8 Hz, 1H, H-6b), 3.88 (dt, J5,6a = J5,6b = 6.8 Hz, J5,4 = 1.0 Hz, 1H, H-5), 3.65 (s, 3H, –OCH3), 3.59–3.47 (m, 1H, H-12′), 2.43–2.24 (m, 4H, 2 × H-11′, 2 × H-2′), 2.13 (s, 3H, –OCOCH3), 2.02 (s, 3H, –OCOCH3), 2.02 (s, 3H, –OCOCH3), 2.00–1.95 (m, 5H, 2 × H-8′, –OCOCH3), 1.66–1.52 (m, 2H, 2 × H-7′), 1.48–1.37 (m, 2H, 2 × H-13′), 1.37–1.14 (m, 16H, –(CH2)8–), 0.86 (t, J18′17′ = 6.9 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.37 (C-1′), 170.48, 170.46, 170.31, 169.36 (4 × OCOCH3), 132.03 (C-9′), 125.08 (C-10′), 101.82 (C-1), 82.27 (C-12′), 71.21 (C-3), 70.61 (C-5), 69.34 (C-2), 67.20 (C-4), 61.46 (C-6), 51.54 (–OCH3), 34.19 (C-13′), 34.02 (C-2′), 33.41 (C-11′), 31.98, 29.62, 29.54, 29.29, 29.26, 29.22, 27.56, 25.31, 25.04, 22.80, 22.75 (m, –(CH2)10–), 20.88, 20.81, 20.76, 20.72 (4 × OCOCH3), 14.18 (C-18′); HR MS m/z calculated for C33H54 O12 [M + Na]+ = 665.3513, found 665.3519.
Methyl 12-O-(β-D-galactopyranosyl)-ricinoleate (22). The title compound was prepared from 21 (200 mg, 0.31 mmol) by the general procedure described above. The reaction time was 0.5 h; and 22 (135 mg) was obtained in 92% yield as a thick syrup. [α]23D = −9.8° (c = 1, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.52–5.39 (m, 2H, H-9′, H-10′), 4.31 (d, J1,2 = 7.1 Hz, 1H, H-1), 3.87 (d, J4,3 = J4,5 = 2.8 Hz, 1H, H-4), 3.80–3.69 (m, 3H, H-6a, 6b, H-12′), 3.67 (s, 3H, OCH3), 3.56–3.45 (m, 3H, H-2, H-3, H-5), 2.49–2.28 (m, 4H, 2 × H-11′, 2 × H-8′), 2.14–2.00 (m, 2H, 2 × H-2′), 1.67–1.58 (m, 2H, H-7′), 1.58–1.50 (m, 2H, 2 × H-13′), 1.50–1.27 (m, 16H, –(CH2)8–), 0.92 (t, J18′17′ = 6.6 Hz 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 175.99 (C-1′), 132.27 (C-9′), 126.95 (C-10′), 104.81 (C-1), 81.10 (C-12′), 76.35 (C-3), 75.04 (C-2), 72.78 (C-5), 70.12 (C-4), 62.22 (C-6), 51.98 (–OCH3), 34.80 (C-13′), 34.23 (C-8′), 33.02 (C-11′), 30.67, 30.29, 30.22, 30.17, 28.41, 26.14, 26.02, 23.72 (m, –(CH2)10–), 14.47 (C-18′); HR MS m/z calculated for C25H46 O8 [M + Na]+ = 497.3090, found 497.3110.
12-O-(β-D-Galactopyranosyl)-ricinoleic acid (23). The title compound was prepared from 21 (300 mg, 0.46 mmol) by the general procedure described above. The reaction time was 0.5 h; and 23 (185 mg) was obtained in 87% yield as a thick syrup. [α]23D = −9.9° (c = 0.3, MeOH); 1H NMR (400 MHz, DMSO) δ 5.37 (m, 2H, H-9′, H-10′), 4.11 (d, J1,2 = 4.5 Hz, 1H, H-1), 3.85–3.33 (m, 4H, H-4, H-12′, H-6a,b), 3.33–3.16 (m, 3H, H-5, H-3, H-2), 2.38–2.07 (m, 4H, 2 × H-11′, 2 × H-2′), 1.97 (m, 2H, 2 × H-8′), 1.56–1.03 (m, 20H, –(CH2)10–), 0.84 (t, J18′,17′ = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO) δ 174.83 (C-1′), 131.11 (C-9′), 126.18 (C-10′), 103.72 (C-1), 78.92 (C-12′), 75.04 (C-5), 73.68 (C-2), 70.96 (C-3), 68.12 (C-4), 60.38 (C-6), 33.87 (C-2′), 33.51 (C-11′), 33.15 (C-13′), 31.51, 29.21, 29.15, 28.86, 28.77, 28.75, 27.04, 24.69, 24.54, 22.28 (m, –(CH2)8–) 14.19 (C-18′); HR MS m/z calculated for C24H44 O8 [M + Na]+ = 483.2934, found 483.2941.
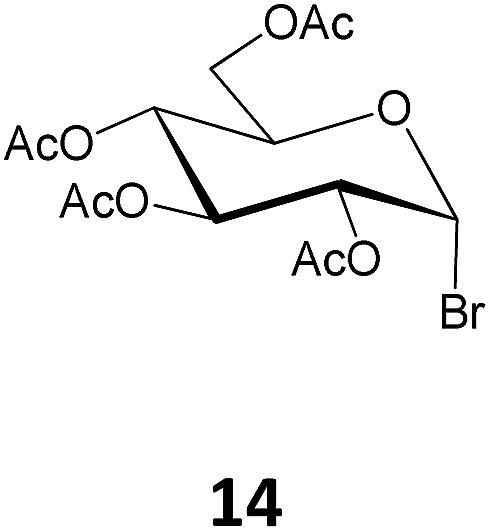
Methyl 12-O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-ricinoleate (24). The title compound was prepared from 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide (14, 500 mg, 1.2 mmol) by the general procedure described above. The reaction time was 14 h; and 24 (455 mg) was obtained in 58% yield as a thick syrup. [α]23D = +10.1° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.47–5.38 (m, 1H, H-9′), 5.35 (m, 1H, H-10′), 5.19 (t, J3,2 = J3,4 = 9.5 Hz, 1H, H-3), 5.06 (t, J4,5 = J4,3 = 9.7 Hz, 1H, H-4), 4.97 (dd, J2,3 = 9.6 Hz, J2,1 = 8.0 Hz, 1H, H-2), 4.55 (d, J1,2 = 8.0 Hz, 1H, H-1), 4.24 (dd, J6a,6b = 12.2 Hz, J6a,5 = 5.2 Hz, 1H, H-6a), 4.11 (dd, J6b,6a = 12.1 Hz, J6b,5 = 2.5 Hz, 1H, H-6b), 3.72–3.66 (m, 1H, H-5), 3.66 (s, 3H, –OCH3), 3.59–3.49 (m, 1H, H-12′), 2.47–2.18 (m, 4H, 2 × H-11′, 2 × H-2′), 2.12–1.92 (m, 14H, 4 × OCOCH3, 2 × H-8′), 1.67–1.53 (m, 2H, 2 × H-7′), 1.51–1.36 (m, 2H, 2 × H-13′), 1.38–1.11 (m, 16H, –(CH2)8–), 0.87 (t, J18′17′ = 6.9 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.44 (C-1′), 170.80, 170.53, 169.57, 169.34 (4 × OCOCH3), 132.09 (C-9′), 125.06 (C-10′), 101.22 (C-1′), 82.10 (C-12′), 73.11 (C-3), 71.78 (C-2), 71.72 (C-5), 68.76 (C-4), 62.34 (C-6), 51.59 (–OCH3), 34.22 (C-13′), 34.03 (C-2′), 33.35 (C-11′), 32.00, 29.65, 29.54, 29.32, 29.28, 29.25, 27.57, 25.31, 25.07, 22.77 (m, –(CH2)8–), 20.86, 20.80, 20.79, 20.76 (4 × OCOCH3), 14.21 (C-18′); HR MS m/z calculated for C33H54 O12 [M + Na]+ = 665.3513, found 665.3519.
The orthoester 9a formed as the by-product in the above reaction was also isolated (132 mg, 17%). 1H NMR (400 MHz, CDCl3) δ 5.59–5.45 (m, 1H, H-9′), 5.42–5.30 (m, 1H, H-10′), 5.18 (t, J3,4 = J3,2 = 9.7 Hz, 1H, H-3), 5.02 (d, J1,2 = 4.0 Hz, 1H, H-1), 4.99 (d, J4,3 = J4,5 = 9.8 Hz, 1H, H-4), 4.26 (dd, J6a,6b = 12.3 Hz, J6a,5 = 4.8 Hz, 1H H-6a), 4.11–4.00 (m, 2H, H-6b, H-5), 3.71–3.59 (m, 5H, H-2, H-12′, –OCH3), 2.38–2.22 (m, 4H, 2 × H-11′, 2 × H-2′), 2.13–1.98 (m, 11H, 3 × OCOCH3, 2 × H-8′), 1.70–1.47 (m, 7H, 2 × H-7′, 2 × H-13′, –CH3), 1.44–1.14 (m, 16H, –(CH2)8–), 0.89 (t, J18′17′ = 7.0 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.34 (C-1′), 170.97, 170.69, 169.67 (3 × OCOCH3), 133.28 (C-9′), 124.64 (C-10′), 97.84 (C-1), 79.95 (C-12′), 77.23 (O–C–O), 73.51 (C-3), 70.95 (C-2), 68.06 (m, C-4, C-5), 62.11 (C-6), 51.48 (–OCH3), 34.76 (–CH3), 34.10 (C-13′), 31.79 (C-2′), 31.45 (C-11′), 29.42, 29.38, 29.13, 29.10, 27.39, 25.62, 24.94, 22.63 (m, –(CH2)8–), 20.93, 20.74, 20.69 (4 × OCOCH3), 14.09 (C-18′); MALDI MS m/z calculated for C33H54 O12 [M + K]+ = 681.873, found 681.747.
Methyl 12-O-(β-D-glucopyranosyl)-ricinoleate (25). The title compound was prepared from 24 (150 mg, 0.23 mmol) following the general procedure described above. The reaction time was 0.5 h; and 25 (98 mg) was obtained in 90% yield as a thick syrup. [α]23D = −21.4° (c = 0.8, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.51–5.40 (m, 2H, H-9′, H-10′), 4.34 (d, J1,2 = 7.8 Hz, 1H, H-1), 3.86 (dd, J6a,6b = 11.8 Hz, J6a,5 = 2.3 Hz, 1H, H-6a), 3.75–3.67 (m, 2H, H-12′, H-6b), 3.66 (s, 3H, –OCH3), 3.39–3.34 (m, 2H, H-3, H-4), 3.28–3.23 (m, 1H, H-5), 3.18 (t, J1,2 = J2,3 = 8.4 Hz, 1H, H-2), 2.49–2.26 (m, 4H, 2 × H-11′, 2 × H-2′), 2.13–2.02 (m, 2H, 2 × H-8′), 1.67–1.58 (m, 2H, 2 × H-7′), 1.57–1.44 (m, 2H, 2 × H-13′), 1.44–1.22 (m, 16H, –(CH2)8–), 0.91 (t, J18′17′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 176.01 (C-1′), 132.35 (C-9′), 126.89 (C-10′), 104.20 (C-1), 81.14 (C-12′), 78.14 (C-3), 77.76 (C-5), 75.34 (C-2), 71.69 (C-4), 62.82 (C-6), 51.98 (–OCH3), 34.80 (C-13′), 34.79 (C-2′), 34.20 (C-11′), 33.03, 30.69, 30.66, 30.30, 30.24, 30.18, 28.42, 26.14, 26.02, 23.72 (m, –(CH2)10–) 14.46 (C-18′); HR MS m/z calculated for C25H46 O8 [M + Na]+ = 497.3090, found 497.3098.
12-O-(β-D-Glucopyranosyl)-ricinoleic acid (26). The title compound was prepared from 24 (250 mg, 0.39 mmol) following the general procedure described above. Reaction time was 3 h; and 26 (145 mg) was obtained in 81% yield as a thick syrup. [α]23D = −14.8° (c = 0.3, MeOH); 1H NMR (400 MHz, CD3OD) δ 5.51–5.35 (m, 2H, H-9′, H-10′), 4.32 (d, J1,2 = 7.8 Hz, 1H, H-1), 3.84 (dd, J6a,6b = 11.8 Hz, J6a,5 = 2.2 Hz, 1H, H-6a), 3.74–3.61 (m, 2H, H-12′, H-6b), 3.38–3.30 (m, 2H, H-3, H-4), 3.27–3.21 (m, 1H, H-5), 3.16 (t, J1,2 = J2,3 = 8.3 Hz, 1H, H-2), 2.47–2.22 (m, 4H, m, 4H, 2 × H-11′, 2 × H-2′), 2.10–1.99 (m, 2H, 2 × H-8′), 1.64–1.55 (m, 2H, 2 × H-7′), 1.55–1.47 (m, 2H, 2 × H-13′), 1.48–1.24 (m, 16H, –(CH2)8–), 0.89 (t, J18′17′ = 6.6 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 178.05 (C-1′), 132.37 (C-9′), 126.86 (C-10′), 104.17 (C-1), 81.13 (C-12′), 78.11 (C-3), 77.72 (C-5), 75.31 (C-2), 71.67 (C-4), 62.80 (C-6), 35.26 (C-13′), 34.77 (C-2′), 34.18 (C-11′), 32.92, 30.71, 30.65, 30.35, 30.27, 28.43, 26.20, 26.12, 23.71 (m, –(CH2)10–), 14.47 (C-18′); HR MS m/z calculated for C24H44 O8 [M + Na]+ = 483.2934, found 483.2934.
Lithium 12-O-(β-D-glucopyranosyl)-ricinoleate (27). The title compound was prepared from 24 (250 mg, 0.39 mmol) by the general procedure described above using LiOH as the alkali. The reaction time was 3 h; and 27 was obtained as colourless fluffy solid (152 mg) in 83% yield. [α]23D = −8.4° (c = 0.5, H2O); 1H NMR (400 MHz, D2O) δ 5.56–5.32 (m, 2H, H-9′, H-10′), 4.37 (d, J1,2 = 7.7 Hz, 1H, H-1), 3.86–3.72 (m, 2H, H-6a,6b), 3.71–3.58 (m, 1H, H-12′), 3.51–3.39 (m, 2H, H-3, H-4), 3.34–3.18 (m, 2H, H-5, H-2), 2.53–2.20 (m, 2H, 2 × H-11′), 2.14 (t, J2′,3′ = 7.5 Hz 2H, 2 × H-2′), 2.08–1.95 (m, 2H, 2 × H-8′), 1.59–1.45 (m, 4H, 2 × H-7′, 2 × H-13′), 1.41–1.18 (m, 16H, –(CH2)8–) 0.84 (t, J18′,17′ = 6.5 Hz, 3H, –CH3); 13C NMR (100 MHz, D2O) δ 181.50 (C-1′), 132.55 (C-9′), 125.06 (C-10′), 102.55 (C-1), 80.89 (C-12′), 75.96 (C-3), 75.71 (C-5), 73.23 (C-2), 69.27 (C-4), 60.54 (C-6), 37.81 (C-2′), 33.31 (C-13′), 32.61 (C-11′), 31.60, 29.32, 29.28, 29.14, 29.01, 28.95, 27.23, 26.14, 24.84, 23.24, 22.48 (m, –(CH2)10–), 13.89 (C-18′); HR MS m/z calculated for C24H43LiO8 [M + Na]+ = 489.3016, found 489.3011.
Methyl 12-O-(2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl)-ricinoleate (28). The title compound was prepared from 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl bromide (15, 1.00 g, 2.4 mmol) following the general procedure described above. The reaction time was 14 h; and 28 (0.92 mg) was obtained in 60% yield as a thick syrup. [α]23D = +51.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.53–5.41 (m, 1H, H-9′), 5.38–5.21 (m, 3H, H-3, H-10′ H-4), 5.17 (dd, J2,3 = 3.3 Hz, J2,1 = 1.8 Hz, 1H, H-2), 4.94 (d, J1,2 = 1.6 Hz, 1H, H-1), 4.27 (dd, J6a,6b = 12.7 Hz, J6a,5 = 5.8 Hz, 1H, H-6a), 4.11–4.02 (m, 2H, H-6b, H-5), 3.66 (s, 3H, –OCH3), 3.65–3.60 (m, 1H, H-12′), 2.33–2.19 (m, 4H, 2 × H-2′, 2 × H-11′), 2.15 (s, 3H, –OCOCH3), 2.09 (s, 3H, –OCOCH3), 2.04 (s, 3H, –OCOCH3), 2.02–1.97 (m, 5H, –OCOCH3, 2 × H-8′), 1.67–1.46 (m, 4H, 4H, 2 × H-7′, 2 × H-13′), 1.42–1.18 (m, 16H, –(CH2)8–), 0.88 (t, J18′,17′ = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.43 (C-1′), 170.77, 170.19, 170.00, 169.91 (4 × OCOCH3), 132.85 (C-9′), 124.32 (C-10′), 96.48 (C-1), 78.81 (C-12′), 70.30 (C-2), 69.30 (C-3), 68.91 (C-5), 66.43 (C-4), 62.74 (C-6), 51.57 (–OCH3), 34.52 (C-13′), 34.24 (C-8′), 31.91, 31.34, 29.61, 29.45, 29.29, 29.25, 27.52, 25.83, 25.08, 22.76, 21.08, 20.87, 20.85 (m, –(CH2)10–), 14.22 (C-18′); HR MS m/z calculated for C33H54 O12 [M + Na]+ = 665.3513, found 665.3550.
Methyl 12-O-(α-D-mannopyranosyl)-ricinoleate (29). The title compound was prepared from 28 (0.5 g, 0.78 mmol) by the general procedure described above. The reaction time was 0.5 h; and compound 29 (0.35 g) was obtained in 94% yield as a thick syrup. [α]23D = +49.8° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.56–5.45 (m, 1H, H-9′), 5.45–5.36 (m, 1H, H-10′), 4.92 (s, 1H, H-1) 3.84–3.59 (m, 10H, H-2, H-6a,b, H-12′ H-3, H-4, H-5, –OCH3), 2.39–2.23 (m, 4H, 2 × H-2′, 2 × H-11′), 2.13–2.03 (m, 2H, 2 × H-8′), 1.68–1.57 (m, 2H, 2 × H-7′), 1.58–1.49 (m, 2H, H-13′), 1.47–1.26 (m, 16H), 0.92 (t, J17′,18′ = 6.5 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 175.98 (C-1′), 133.03 (C-9′), 126.07 (C-10′), 99.91 (C-1), 77.59 (C-12′), 74.84 (C-4), 72.67 (C-2), 72.64 (C-3), 68.45 (C-5), 62.79 (C-6), 51.98 (–OCH3), 35.68 (C-13′), 34.79 (C-11′), 32.98, 31.82, 30.64, 30.54, 30.26, 30.21, 30.15, 28.37, 26.69, 26.01, 23.73 (m, –(CH2)10–), 14.48 (C-18′); HR MS m/z calculated for C25H46 O8 [M + Na]+ = 497.3091, found 497.3091.
12-O-(α-D-Mannopyranosyl)-ricinoleic acid (30). The title compound was prepared from 28 (300 mg, 0.46 mmol) by the general procedure described above. The reaction time was 3 h; and compound 30 (180 mg) was obtained as a thick syrup in a yield of 85%. [α]23D = +68.4° (c = 0.5, MeOH); 1H NMR (400 MHz, DMSO) δ 5.50–5.21 (m, 2H, H-9′, H-10′), 4.73 (s, 1H, H-1), 3.85–3.00 (m, 7H, H-2, H-3, H-4, H-5, H-6a,b, H-12′), 2.18 (m, 4H, 2 × H-11′, 2 × H-2′), 1.99 (m, 2H, 2 × H-8′), 1.58–1.01 (m, 20H, –(CH2)10–), 0.85 (t, J18′,17′ = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO) δ 174.63 (C-1′), 131.61 (C-9′), 125.18 (C-10′), 97.89 (C-1), 74.38 (C-12′), 74.31 (C-4), 71.04 (C-2), 70.89 (C-3), 66.85 (C-5), 61.23 (C-6), 33.99 (C-2′), 33.73 (C-11′), 31.36 (C-13′), 30.22, 29.08, 28.87, 28.74, 28.66, 28.62, 26.90, 25.06, 24.57, 22.18 (m, –(CH2)10–) 14.06 (C-18′); HR MS m/z calculated for C24H44 O8 [M + Na]+ = 483.2934, found 483.2938.
Sodium 12-O-(α-D-mannopyranosyl)-ricinoleate (31). The title compound was prepared from 28 (200 mg, 0.31 mmol) by the general procedure described above using NaOH as the alkali. The reaction time was 3 h; and compound 31 (120 mg) was obtained in 80% yield as a colourless fluffy solid. [α]23D = +45.9° (c = 0.5, H2O); 1H NMR (400 MHz, D2O) δ 5.62–5.46 (m, 1H, H-9′), 5.46–5.28 (m, 1H, H-10′), 4.98 (s, 1H, H-1), 3.90–3.82 (m, 2H, H-2, H-6a), 3.82–3.57 (m, 5H, H-3, H-6a, H-12′, H-5, H-4), 2.37–2.12 (m, 4H, 2 × H-11′, 2 × H-2′), 2.12–1.99 (m, 2H, 2 × H-8′), 1.69–1.46 (m, 4H, 2 × H-7′, 2 × H-13′), 1.46–1.14 (m, 16H, –(CH2)8–), 0.87 (t, J17′,18′ = 6.3 Hz, 3H, –CH3); 13C NMR (100 MHz, D2O) δ 183.42 (C-1′), 132.82 (C-9′), 124.39 (C-10′), 98.36 (C-1), 76.54 (C-12′), 72.95 (C-4), 70.81 (C-2), 70.76 (C-3), 66.10 (C-5), 60.43 (C-6), 37.71 (C-2′), 34.14 (C-13′), 31.54, 30.43, 29.31, 29.23, 29.14, 28.99, 28.91, 27.20, 26.11, 25.38, 22.49 (m, –(CH2)11–), 13.85 (C-18′); HR MS m/z calculated for C24H43NaO8 [M + Na]+ = 505.2753, found 505.2751.
Methyl 12-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl)-ricinoleate (32). The title compound was prepared from 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-D-glucopyranosyl chloride (16, 1.00 g, 2.74 mmol) following the general procedure described above. The reaction time was 16 h; and 32 (930 mg) was obtained as a colorless glassy solid in 53%. [α]23D = −15.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.61 (brs, 1H, –NH), 5.47–5.28 (m, 3H, H-9′, H-10′, H-3), 5.02 (t, J4,3 = J4,5 = 9.6 Hz, 1H, H-4), 4.78 (d, J1,2 = 8.3 Hz, 1H, H-1), 4.23 (dd, J6a,6b = 12.1 Hz, J6a,5 = 5.2 Hz, 1H, H-6a), 4.10 (dd, J6b,6a = 12.1 Hz, J6b,5 = 1.9 Hz, 1H), 3.78–3.67 (m, 2H, H-2, H-5), 3.65 (s, 3H, –OCH3), 3.57–3.46 (m, 1H, H-12′), 2.43–2.26 (m, 4H, 2 × H-11′, 2 × H-2′), 2.07–1.89 (m, 14H, 4 × OCOCH3, 2 × H-8′), 1.67–1.53 (m, 2H, 2 × H-7′), 1.48–1.37 (m, 2H, 2 × H-13′), 1.26 (m, 16H, –(CH2)8–) 0.86 (t, J18′,17′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.53 (C-1′), 170.99, 170.84, 170.14, 169.64 (4 × OCOCH3), 132.03 (C-9′), 125.22 (C-10′), 101.15 (C-1), 82.11 (C-12′), 72.42 (C-3), 71.69 (C-5), 69.08 (C-4), 62.56 (C-6), 55.66 (C-2), 51.61 (–OCH3), 34.22 (C-13′), 34.05 (C-2′), 33.35 (C-11′), 32.05, 29.61, 29.56, 29.29, 29.23 (m, –(CH2)8–), 27.54 (C-8′), 25.50 (–(CH2)8–), 25.06 (C-7′), 23.46 (NHCOCH3), 22.79, 20.87, 20.81 (3 × OCOCH3), 14.22 (C-18′); HR MS m/z calculated for C33H55NO12 [M + Na]+ = 664.3673, found 664.3694.
Methyl 12-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-ricinoleate (33). The title compound was prepared from 32 (0.5 g, 0.78 mmol) following the general procedure described above. The reaction time was 0.5 h; 16 h; and 33 (0.46 g) was obtained as a light brown glassy solid in 92% yield. [α]23D = −40.8° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.41 (m, 2H, H-9′, H-10′), 4.49 (d, J1,2 = 8.2 Hz, 1H, H-1), 3.86 (d, J6a,6b = J6a,5 = 11.8 Hz, 1H, H-6a), 3.70 (dd, J6b,6a = 11.8 Hz, J6b,5 = 5.1 Hz, 1H, H-6b), 3.65 (s, 3H, –OCH3), 3.60 (m, 2H, H-12′, H-2), 3.49 (t, J3,2 = J3,4 = 9.3 Hz, 1H, H-3), 3.38–3.22 (m, 2H, H-4, H-5), 2.56–2.20 (m, 4H, 2 × H-11′, 2 × H-2′), 2.05 (m, 2H, 2 × H-8′), 1.97 (s, 3H, NHCOCH3), 1.66–1.54 (m, 2H, 2 × H-7′), 1.51–1.39 (m, 2H, 2 × H-13′), 1.41–1.29 (m, 16H, –(CH2)8–), 0.90 (t, J18′,17′ = 6.2 Hz, 3H, CH3); 13C NMR (100 MHz, CD3OD) δ 176.17 (NHCOCH3), 173.53 (C-1′), 132.52 (C-9′), 126.70 (C-10′), 103.01 (C-1), 82.31 (C-12′), 77.71 (C-5), 75.83 (C-3), 72.00 (C-4), 62.74 (C-6), 57.85 (C-2), 52.06 (–OCH3), 34.81 (C-2′), 34.21 (C-13′), 33.10 (C-11′), 30.72, 30.67, 30.28, 30.22, 30.16, 28.40, 26.17, 26.01, 23.76 (m, –(CH2)8–) 23.16 (NHCOCH3), 14.48 (C-18′); HR MS m/z calculated for C27H49NO8 [M + Na]+ = 538.3356, found 538.3375.
12-O-(2-Acetamido-2-deoxy-β-D-glucopyranosyl)-ricinoleic acid (34). The title compound was prepared from 32 (200 mg, 0.31 mmol) by the general procedure described above. The reaction time was 3 h; and compound 34 (130 mg) was obtained as a light brown fluffy solid in 83% yield. [α]23D = +4.8° (c = 0.25, MeOH); 1H NMR (400 MHz, DMSO) δ 7.86 (d, JNH,2 = 7.2 Hz, 1H, –NH), 5.43–5.26 (m, 2H, H-9′, H-10′), 4.31 (d, J1,2 = 6.9 Hz, 1H, H-1), 3.72–3.51 (m, 2H, H-6a,b), 3.46–3.19 (m, 3H, H-12′, H-2, H-5), 3.17–2.83 (m, 2H, H-3, H-4), 2.46–2.01 (m, 2H, 2 × H-11′), 2.03–1.80 (m, 4H, 2 × H-8′, 2 × H-2′), 1.76 (s, 3H, NHCOCH3), 1.43–1.00 (m, 20H, –(CH2)10–), 0.84 (t, J18′,17′ = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO) δ 168.97 (C-1′), 131.24 (C-9′), 125.90 (C-10′), 102.13 (C-1), 80.25 (C-12′), 76.95 (C-4), 74.04 (C-5), 70.81 (C-3), 61.24 (C-6), 55.91 (C-2), 33.44, 33.11, 31.45, 29.45, 29.22, 29.09, 29.07, 28.86, 26.97, 24.42, 23.10, 22.20 (m, –(CH2)8–), 14.08 (C-18′); HR MS m/z calculated for C26H47NO8 [M + Na]+ = 524.3200, found 524.3193.
Methyl 12-O-(2,3,4-tri-O-benzoyl-α-L-rhamnopyranosyl)-ricinoleate (35). The title compound was prepared from 2,3,4-tri-O-benzoyl-α-L-rhamnopyranosyl bromide (17, 1.00 g, 2.0 mmol) following the general procedure described above. The reaction time was 18 h; and compound 35 (1.016 g) was obtained in 66% yield as a thick syrup. [α]23D = +84.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, JA,B = 8.0 Hz, JA,B = 1.0 Hz, 2H, Ph-CH), 7.98 (dd, JA,B = 8.1, JA,B = 1.0 Hz, 2H, Ph-CH), 7.83 (dd, JA,B = 8.2 Hz, JA,B = 1.0 Hz, 2H, Ph-CH), 7.65–7.55 (m, 1H, Ph-CH), 7.55–7.35 (m, 6H, Ph-CH), 7.26 (dd, JA,B = 9.0 Hz, JA,B = 6.6 Hz, 2H, Ph-CH), 5.83 (dd, J3,4 = 10.1 Hz, J3,2 = 3.4 Hz, 1H, H-3), 5.67 (t, J4,3 = J4,5 = 10.0 Hz, 1H, H-4), 5.60 (dd, J2,3 = 3.2, J2,1 = 1.7 Hz, 1H), 5.58–5.44 (m, 2H, H-9′, H-10′), 5.10 (s, 1H, H-1), 4.33 (dq, J5,CH3 = 12.4 Hz, J5,4 = 6.2 Hz, 1H, H-5), 3.76–3.68 (m, 1H, H-12′), 3.65 (s, 3H, –OCH3), 2.51–2.23 (m, 4H, 2 × H-11′, 2 × H-2′), 2.17–2.01 (m, 2H, 2 × H-8′), 1.71–1.50 (m, 4H, 2 × H-7′, 2 × H-13′), 1.45–1.24 (m, 19H, –(CH2)8–, –CH3), 0.89 (t, J17′,18′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.26 (C-1′), 165.83, 165.64, 165.49 (3 × Ph-CO), 133.39, 133.26
133.03 (3 × Ph-C), 132.25 (C-9′), 129.90, 129.72, 129.68, 129.57, 129.41, 129.32, 128.56, 128.40, 128.25 (m, Ph-C) 125.48 (C-10′), 96.81 (C-1), 79.46 (C-12′), 72.03 (C-4), 71.47 (C-2), 70.13 (C-3), 66.78 (C-5), 51.42 (–OCH3), 34.08 (C-2′), 33.58 (C-13′), 32.73 (C-11′), 31.79, 29.62, 29.45, 29.21, 29.14, 27.51, 25.14, 24.95, 22.65, 22.58, 17.58 (m, –(CH2)10–), 14.11 (C-18′); HR MS m/z calculated for C46H58 O10 [M + Na]+ = 793.3928, found 793.3933.
Methyl 12-O-(α-L-rhamnopyranosyl)-ricinoleate (36). The title compound was prepared from 35 (500 mg, 0.65 mmol) by the general procedure described above. The reaction time was 4 h; and 36 (270 mg) was obtained in 91% yield as a thick syrup. [α]23D = −57.8° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.53–5.34 (m, 2H, H-9′, H-10′), 4.77 (d, J1,2 = 1.4 Hz, 1H, H-1), 3.78 (dd, J2,3 = 3.3 Hz, J2,1 = 1.6 Hz, 1H, H-2), 3.76–3.67 (m, 1H, H-5), 3.67–3.57 (m, 5H, –OCH3, H-3, H-12′), 3.38 (t, J4,3 = J4,5 = 9.5 Hz, 1H, H-4), 2.40–2.21 (m, 4H, 2 × H-11′, 2 × H-2′), 2.11–2.01 (m, 2H, 2 × H-8′), 1.67–1.44 (m, 4H, 2 × H-7′, 2 × H-13′), 1.44–1.26 (m, 16H, –(CH2)8–), 1.24 (d, J5,CH3 = 6.3 Hz, 3H, –CH3), 0.91 (t, J17′,18′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 174.72 (C-1′), 131.42 (C-9′), 125.34 (C-10′), 99.72 (C-1), 78.02 (C-12′), 72.53 (C-4), 71.30 (C-2), 71.06 (C-3), 68.65 (C-5), 50.65 (–OCH3), 33.43 (C-2′), 33.01 (C-13′), 32.35 (C-11′), 31.59, 29.24, 29.16, 28.85, 28.81, 28.76, 26.97, 24.76, 24.62, 22.28 (m, –(CH2)10), 16.55 (–CH3), 13.06 (C-18′); HR MS m/z calculated for C24H46 O7 [M + Na]+ = 481.3142, found 481.3154.
12-O-(α-L-Rhamnopyranosyl)-ricinoleic acid (37). The title compound was prepared from 35 (2.00 g, 2.6 mmol) by the general procedure described above. The reaction time was 8 h; and 37 was obtained (1.00 g) in a yield of 87% as a thick syrup. [α]23D = −14.7° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.51–5.38 (m, 2H, H-9′, H-10′), 4.77 (d, J1,2 = 1.1 Hz, 1H, H-1), 3.78 (dd, J2,3 = 3.1 Hz, J2,1 = 1.5 Hz, 1H, H-2), 3.76–3.69 (m, 1H, H-5), 3.68–3.57 (m, 2H, H-3, H-12′), 3.38 (t, J4,3 = J4,5 = 9.5 Hz, 1H, H-4), 2.41–2.20 (m, 4H, 2 × H-11′, 2 × H-2′), 2.14–1.98 (m, 2H, 2 × H-8′), 1.65–1.45 (m, 4H, 2 × H-7′, 2 × H-13′), 1.45–1.26 (m, 16H, –(CH2)8–), 1.24 (d, J5,CH3 = 6.2 Hz, 3H, –CH3), 0.90 (t, J17′,18′ = 6.7 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 177.78 (C-1′), 132.80 (C-9′), 126.69 (C-10′), 101.09 (C-1), 79.40 (C-12′), 73.92 (C-4), 72.69 (C-2), 72.44 (C-3), 70.02 (C-5), 34.98 (C-2′), 34.38 (C-13′), 33.72 (C-11′), 32.97, 30.63, 30.53, 30.28, 30.22, 30.19, 28.36, 26.14, 26.08, 23.65 (m, –(CH2)10), 17.92 (–CH3), 14.44 (C-18′): HR MS m/z calculated for C24H44 O7 [M + Na]+ = 467.2985, found 467.2985.
Methyl 12-O-(2,3,5-tri-O-benzoyl-β-D-arabinofuranosyl)-ricinoleate (38). The title compound was prepared from 2,3,5-tetra-O-benzoyl-α-D-arabinofuranosyl chloride (18, 2.18 g, 4.5 mmol) by the general procedure described above. The reaction time was 16 h; and 38 (1.95 g) was obtained in a yield of 57% as a thick syrup. [α]23D = −14.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.14–7.94 (m, 6H, Ph-CH), 7.63–7.27 (m, 9H, Ph-CH), 5.62–5.26 (m, 5H, H-3, H-2, H-9′, H-10′, H-1), 4.80 (d, J5a,5b = J5a,4 = 10.8 Hz, 1H, H-5a), 4.73–4.53 (m, 2H, H-5b, H-4), 3.83–3.67 (m, 1H, H-12′), 3.66 (s, 3H, –OCH3), 2.42–2.17 (m, 4H, 2 × H-11′, 2 × H-2′), 2.01–2.00 (m, 2H, H-8′), 1.68–1.16 (20H, –(CH2)10–), 0.86 (t, J17′,18′ = 5.6 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.44 (C-1′), 166.37, 165.97, 165.58 (3 × C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 133.62, 133.57, 133.16, 132.66 (Ph-C), 131.88 (C-9′), 130.09, 130.00, 129.90, 129.34, 129.32, 128.62, 128.59, 128.44 (Ph-C), 125.83 (C-10′), 104.92 (C-1), 82.55 (C-2), 80.94 (C-4), 78.07 (C-3), 77.77 (C-12′), 64.07 (C-5), 51.58 (–OCH3), 34.22 (C-2′), 33.79 (C-11′), 32.85 (C-13′), 31.93, 29.67, 29.59, 29.29, 29.24, 27.52, 25.31, 25.06, 22.75 (–(CH2)10–), 14.23 (C-18′); HR MS m/z calculated for C45H56 O11 [M + Na]+ = 779.3371, found 779.3372.
O), 133.62, 133.57, 133.16, 132.66 (Ph-C), 131.88 (C-9′), 130.09, 130.00, 129.90, 129.34, 129.32, 128.62, 128.59, 128.44 (Ph-C), 125.83 (C-10′), 104.92 (C-1), 82.55 (C-2), 80.94 (C-4), 78.07 (C-3), 77.77 (C-12′), 64.07 (C-5), 51.58 (–OCH3), 34.22 (C-2′), 33.79 (C-11′), 32.85 (C-13′), 31.93, 29.67, 29.59, 29.29, 29.24, 27.52, 25.31, 25.06, 22.75 (–(CH2)10–), 14.23 (C-18′); HR MS m/z calculated for C45H56 O11 [M + Na]+ = 779.3371, found 779.3372.
Methyl 12-O-(β-D-arabinofuranosyl)-ricinoleate (39). The title compound was prepared from 38 (1.00 g, 1.32 mmol) by the general procedure described above. The reaction time was 3 h; and 39 (540 mg) was obtained in 92% yield as a thick syrup. [α]23D = −74.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.50–5.37 (m, 2H, H-9′, H-10′), 4.97 (d, J1,2 = 1.5 Hz, 1H, H-1), 4.00–3.92 (m, 2H, H-4, H-2), 3.84 (dd, J3,2 = 6.4 Hz, J3,4 = 3.7 Hz, 1H, H-3), 3.74 (dd, J5a,5b = 11.9 Hz, J5a,4 = 3.2 Hz, 1H, H-5a), 3.69–3.59 (m, 5H, –OCH3, H-5b, H-12′), 2.38–2.25 (m, 4H, 2 × H-2′, 2 × H-11′), 2.10–2.00 (m, 2H, 2 × H-8′), 1.66–1.44 (m, 4H, 2 × H-7′, 2 × H-13′), 1.44–1.25 (m, 16H, –(CH2)8–), 0.90 (t, J17′,18′ = 6.7 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 176.18 (C-1′), 132.46 (C-9′), 127.00 (C-10′), 108.80 (C-1), 85.22 (C-4), 83.66 (C-2), 78.99 (C-3), 78.65 (C-12′), 62.85 (C-5), 52.05 (–OCH3), 34.81 (C-2′), 34.68 (C-13′), 34.04 (C-11′), 32.97, 30.62, 30.56, 30.22, 30.16, 30.12, 28.34, 26.26, 25.99, 23.67 (m, –(CH2)10–), 14.44 (C-18′); HR MS m/z calculated for C24H44 O7 [M + Na]+ = 467.2985, found 467.3003.
12-O-(β-D-Arabinofuranosyl)-ricinoleic acid (40). The title compound was prepared from 38 (1.00 g, 1.32 mmol) by the general procedure described above. The reaction time was 8 h and compound 40 (0.45 g) was obtained in 79% yield as a thick syrup. [α]23D = −67.0° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.49–5.39 (m, 2H, H-9′, H-10′), 4.97 (d, J1,2 = 1.4 Hz, 1H, H-1), 4.00–3.94 (m, 2H, H-4, H-2), 3.84 (dd, J3,2 = 6.3 Hz, J3,4 = 3.7 Hz, 1H, H-3), 3.74 (dd, J5a,5b = 11.9 Hz, J5a,4 = 3.3 Hz, 1H, H-5a), 3.68–3.59 (m, 2H, H-5b, H-12′), 2.41–2.24 (m, 4H, 2 × H-11′, 2 × H-2′), 2.11–1.98 (m, 2H, 2 × H-8′), 1.67–1.45 (m, 4H, 2 × H-7′, 2 × H-13′), 1.45–1.22 (m, 16H, –(CH2)8–), 0.90 (t, J17′,18′ = 6.7 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 177.89 (C-1′), 132.49 (C-9′), 126.96 (C-10′), 108.75 (C-1), 85.19 (C-4), 83.64 (C-2), 79.01 (C-3), 78.62 (C-12′), 62.81 (C-5), 34.98 (C-2′), 34.65 (C-13′), 34.01 (C-11′), 32.95, 30.62, 30.54, 30.25, 30.18, 30.16, 28.34, 26.24, 26.05, 23.65 (m, –(CH2)10–), 14.44 (C-18′); HR MS m/z calculated for C23H42 O7 [M + Na]+ = 453.2829, found 453.2838.
Methyl 12-O-(2,3,4,6-tetra-O-acetyl-β-D-galactofuranosyl)-ricinoleate (41). The title compound was prepared from 2,3,4,6-tetra-O-acetyl-α-D-galactofuranosyl chloride (19, 2.00 g, 5.5 mmol) by the general procedure described above. The reaction time was 14 h; and 41 (2.12 g) was obtained in 60% yield as a thick syrup. [α]23D = −24.4° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.48–5.32 (m, 3H, H-9′, H-10′, H-5), 5.08 (s, 1H, H-1), 5.00 (dd, J2,3 = 2.0 Hz, J1,2 = 0.5 Hz, 1H, H-2), 4.96 (dd, J3,4 = 6.1 Hz, J3,2 = 1.7 Hz, 1H, H-3), 4.33–4.25 (m, 2H, H-6a, H-4), 4.18 (dd, J6a,6b = 11.7 Hz, J6b,5 = 7.3 Hz, 1H, H-6b), 3.65 (s, 3H, –OCH3), 3.63–3.53 (m, 1H, H-12′), 2.32–2.18 (m, 4H, 2 × H-2′, 2 × H-11′), 2.12, 2.09, 2.06, 2.03 (4 × s, 12H, 4 × OCOCH3), 2.03–1.92 (m, 2H, 2 × H-8′), 1.66–1.54 (m, 2H, 2 × H-7′), 1.52–1.40 (m, 2H, 2 × H-13′), 1.39–1.17 (m, 16H, –(CH2)8–), 0.86 (t, J17′,18′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.38 (C-1′), 170.61, 170.17, 170.16, 169.79 (4 × OCOCH3), 131.96 (C-9′), 125.70 (C-10′), 104.87 (C-1), 81.89 (C-2), 79.73 (C-4), 78.21 (C-12′), 76.64 (C-3), 69.33 (C-5), 62.85 (C-6), 51.56 (–OCH3), 34.20 (C-2′), 33.74 (C-13′), 32.87 (C-11′), 31.90, 29.67, 29.49, 29.30, 29.29, 29.23, 27.54, 25.23, 25.05, 22.72, 20.94, 20.92, 20.81 (m, –(CH2)10–), 14.20 (C-18′); HR MS m/z calculated for C33H54 O12 [M + Na]+ = 665.3513, found 665.3547.
Methyl 12-O-(β-D-galactofuranosyl)-ricinoleate (42). The title compound was prepared from 41 (250 mg, 0.38 mmol) following the general procedure described above. The reaction time was 0.5 h; and 42 (170 mg) was obtained in 94% yield as a thick syrup. [α]23D = −36.6° (c = 0.5, CHCl3); 1H NMR (400 MHz, CD3OD) δ 5.49–5.39 (m, 2H, H-9′, H-10′), 4.95 (d, J1,2 = 1.5 Hz, 1H, H-1), 4.00 (dd, J3,4 = 6.5 Hz, J4,5 = 4.1 Hz, 1H, H-4), 3.95–3.93 (m, 2H, H-2, H-3), 3.72 (m, 1H, H-5), 3.64–3.56 (m, 6H, –OCH3, H-12′, H-6a,b), 2.41–2.23 (m, 4H, 2 × H-2′, 2 × H-11′), 2.12–1.97 (m, 2H, 2 × H-8′), 1.67–1.52 (m, 2H, 2 × H-7′), 1.55–1.42 (m, 2H, 2 × H-13′), 1.42–1.22 (m, 16H, –(CH2)8–), 0.90 (t, J17′,18′ = 6.7 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 176.13 (C-1′), 132.45 (C-9′), 127.06 (C-10′), 108.90 (C-1), 84.11 (C-2), 83.62 (C-3) 79.26 (C-6), 78.63 (C-4), 72.25 (C-5), 64.89 (C-12′), 52.03 (OCH3), 34.81 (C-2′), 34.07 (C-13′), 32.98 (C-11′), 30.65, 30.57, 30.25, 30.18, 30.14, 28.37, 26.29, 26.00, 23.68 (m, –(CH2)10–), 14.45 (C-18′); HR MS m/z calculated for C25H46 O8 [M + Na]+ = 497.3091, found 497.3111.
12-O-(β-D-Galactofuranosyl) ricinoleic acid (43). The title compound was prepared from 41 (300 mg, 0.46 mmol) following the general procedure described above. The reaction time was 3 h; and 43 (181 mg) was obtained in 85% yield as a thick syrup. [α]23D = −31.2° (c = 0.5, CHCl3); 1H NMR (400 MHz, DMSO) δ 5.37 (m, 2H, H-9′, H-10′), 4.74 (s, 1H, H-1), 3.91–3.24 (m, 7H, H-4, H-2, H-3, H-5, H-12′ H-6a,b), 2.34–2.07 (m, 2H, H-11′), 2.05–1.84 (m, 4H, 2 × H-2′, 2 × H-8′), 1.58–1.09 (m, 20H, –(CH2)10–), 0.84 (t, J18′,17′ = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO) δ 174.46 (C-1′), 131.07 (C-9′), 126.18 (C-10′), 107.56 (C-1), 82.56 (C-2), 82.06 (C-3), 77.00 (C-12′), 76.74 (C-4), 70.48 (C-5), 63.36 (C-6), 36.84 (C-2′), 33.53 (C-13′), 32.89 (C-11′), 31.43, 29.23, 29.03, 28.82, 27.01, 25.82, 24.80, 22.23 (m, –(CH2)10–) 14.12 (C-18′); HR MS m/z calculated for C24H44 O8 [M + Na]+ = 483.2934, found 483.2933.
Methyl 12-O-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-D-lactosyl)-ricinoleate (44). The title compound was prepared from acetobromolactose (20, 5.00 g, 7.15 mmol) following the general procedure described above. The reaction time was 16 h; and 44 (3.52 g) was obtained in 53% yield as a thick syrup. [α]23D = −15.6° (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.44–5.36 (m, 1H, H-9′′), 5.36–5.26 (m, 2H, H-10′′, H-4′), 5.17 (t, J3,4 = J3,2 = 9.3 Hz, 1H, H-3), 5.09 (dd, J2′,3′ = 10.4 Hz, J2′,1′ = 7.9 Hz, 1H, H-2′), 4.94 (dd, J3′,2′ = 10.4 Hz, J3′,4′ = 3.4 Hz, 1H, H-3′), 4.87 (dd, J2,3 = 9.6 Hz, J2,1 = 8.0 Hz, 1H, H-2), 4.50 (d, J1,2 = 8.0 Hz, 1H, H-1), 4.47 (m, (J1′,2′ = 8.0 Hz, H-1′), 2H, H-1′, H-6′a), 4.08 (m, 3H, H-6a,b, H-6′b), 3.86 (m, 1H, H-5′), 3.75 (t, J4,3 = J4,5 = 9.4 Hz, 1H, H-4), 3.65 (s, 3H, OCH3), 3.58 (m, 1H, H-5), 3.54–3.44 (m, 1H, H-12′′), 2.36–2.18 (m, 4H, 2 × H-2′′, 2 × H-11′′), 2.15–1.92 (m, 23H, 7 × –OCOCH3, 2 × H-8′′), 1.59 (m, 2H, 2 × H-7′′), 1.38 (m, 2H, 2 × H-13′′), 1.33–1.15 (m, 16H, –(CH2)8–), 0.86 (t, J18′′,17′′ = 6.9 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.30 (C-1′′), 170.37, 170.34, 170.17, 170.08, 169.89, 169.52, 169.11 (7 × OCOMe), 131.86 (C-9′′), 124.98 (C-10′′), 101.09 (C-1), 100.85 (C-1′), 81.89, (C-12′′) 76.56 (C-4), 73.03 (C-3), 72.44 (C-5), 72.01 (C-2), 70.99 (C-3′), 70.66 (C-5′), 69.11 (C-2′), 66.62 (C-4′), 62.15 (C-6′), 60.82 (C-6), 51.46 (COOCH3), 34.08 (C-13′′), 33.94 (C-8′′), 33.18 (C-11′′), 31.86, 29.50, 29.39, 29.18, 29.14, 29.11, 27.42, 25.19, 24.93, 22.63 (m, –(CH2)8–), 20.83, 20.81, 20.70, 20.64, 20.52 (m, 7 × OCOCH3), 14.07 (C-18′′); HR MS m/z calculated for C45H70 O20 [M + Na]+ = 953.4358, found 953.4364.
Methyl 12-O-(β-D-lactosyl)-ricinoleate (45). The title compound was prepared from 44 (500 mg, 0.54 mmol) following the general procedure described above. The reaction time was 1 h; and 45 (310 mg) was obtained in 91% as a thick syrup. [α]23D = +68.4° (c = 0.5, MeOH); 1H NMR (400 MHz, CD3OD) δ 5.51–5.37 (m, 2H, H-9′′, H-10′′), 4.38 (d, J1,2 = 7.4 Hz, 1H, H-1), 4.37 (d, J1′,2′ = 7.8 Hz, 1H, H-1′) 3.89–3.36 (m, 15H, H-3′, H-4′, H-5′, H-6′a,b, H-2, H-3, H-4, H-5, H-6a,b, H-12′′, –OCH3), 3.24 (dd, J2′,3′ = 8.9 Hz, J2′,1′ = 8.0 Hz, 1H, H-2′), 2.32 (m, 4H, 2 × H-2′′, 2 × H-11′′), 2.06 (m, 2H, H-8′′), 1.63–1.56 (m, 2H, 2 × H-7′′), 1.56–1.47 (m, 2H, 2 × H-13′′), 1.42–1.20 (m, 16H, –(CH2)8–), 0.90 (t, J18′′,17′′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 176.12 (C-1′′), 132.47 (C-9′′), 126.86 (C-10′′), 105.09 (C-1), 104.16 (C-1′), 81.43, 80.78, 77.11, 76.49, 76.33, 74.94, 74.78, 72.60, 70.32, 62.51, 62.09 (m), 52.07 (-OCH3), 34.84, 34.23, 33.05, 30.71, 30.68, 30.32, 30.3, 30.20, 28.45, 26.15, 26.05, 23.75 (m, –(CH2)8–), 14.52 (C-18′′); HR MS m/z calculated for C31H56O13 [M + Na]+ = 659.3619, found 659.3622.
12-O-(β-D-Lactosyl)-ricinoleic acid (46). The title compound was prepared from 44 (200 mg, 0.21 mmol) following the general procedure described above. The reaction time was 4 h; and 46 (110 mg) was obtained in 84% yield as a thick syrup. [α]23D = +39.2° (c = 0.5, MeOH); 1H NMR (400 MHz, CD3OD) δ 5.49–5.41 (m, 2H, H-9′′, H-10′′) 4.41 (d, J1,2 = 7.0 Hz, 1H, H-1), 4.39 (d, J1′,2′ = 7.7 Hz, 1H, H-1′), 3.92–3.77 (m, 4H, H-6a′, H-6b′, H-4′, H-6a), 3.76–3.67 (m, 2H, H-6b, H-12′′), 3.66–3.49 (m, 5H, H-4, H-5, H-3′, H-2, H-3), 3.47–3.39 (m, 1H, H-5′), 3.27 (t, J2′,3′ = J2′,1′ = 8.0 Hz, 1H, H-2′), 2.50–2.19 (m, 4H, 2 × H-11′′, 2 × H-2′′), 2.13–2.00 (m, 2H, H-8′′), 1.68–1.43 (m, 4H, 2 × H-7′′, 2 × H-13′′), 1.44–1.23 (m, 16H, –(CH2)8–), 0.91 (t, J18′′,17′′ = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CD3OD) δ 173.99 (C-1′′), 131.23 (C-9′′), 125.28 (C-10′′), 103.61 (C-1), 102.70 (C-1′), 80.15 (C-12′), 79.28 (C-4), 75.65 (C-5), 75.06 (C-2′), 74.90 (C-5′), 73.51 (C-2), 73.36 (C-3), 71.20 (C-3′), 68.89 (C-4′), 61.09 (C-6), 60.62 (C-6′), 36.05 (C-2′′), 33.37 (C-13′′), 32.76 (C-11′′), 31.58, 29.34, 29.20, 29.06, 28.96, 27.07, 25.66, 24.70, 22.30 (m, –(CH2)8–), 13.10 (C-18′′); HR MS m/z calculated for C30H54O13 [M + Na]+ = 645.3462, found 645.3470.
Methyl 9,10-dibromo-12-O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-ricinoleate (47). Bromine (0.05 mL, 0.87 mmol) was added to a stirred solution of 24 (370 mg, 0.58 mmol) in chloroform (5 mL) at room temperature and the stirring was continued for 5 h at which time TLC showed complete consumption of 24. The reaction mixture was diluted with CH2Cl2 (100 mL) and was washed successively water, aqueous NaHCO3, and brine solutions in the cold. The organic layer was then dried over anhydrous Na2SO4 and was concentrated under reduced pressure to yield, after chromatography on a column of silica gel (eluent, EtOAc
:hexane), the title compound 47 (370 mg, 80%) (as a pair of enantiomers) in the form of a thick syrup. [α]23D = +41.6° (c = 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) (the two enantiomers are marked A & B) δ 5.45 (dd, 2H, J3,4 = 9.5 Hz, J4,3 = 9.9 Hz, H-3A, H-3B), 5.17–5.13 (2 × d, J1,2 = 5.1 Hz, 2H, H-1A, H-1B), 5.06 (2 × t, J4,5 = J4,3 = 10.0 Hz, 2H, H-4A, H-4B), 4.97–4.87 (2 × d, J2,3 = 10.4 Hz, J2,1 = 3.7 Hz, 2H, H-2A, H-2B), 4.29–4.03 (m, 10H, 2 × H-6a, 2 × H-5, 2 × H-9′, 2 × H-10′, 2 × H-6b), 3.85–3.76 (m, 2 × H-12′), 3.66 (s, 6H, 2 × –OCH3), 2.30 (t, 4H, J2′,3′ = 7.5 Hz, 4 × H-2′), 2.1–2.0 (7 × s, 24H, 8 × OCOCH3), 1.67–1.57 (m, 48H, 2 × –(CH2)12), 0.88 (t, J18′,17′ = 6.8 Hz, 6H, 2 × CH3); 13C NMR (100 MHz, CDCl3) δ 174.38 (2 × C-1′), 170.80, 170.35, 170.29, 170.18, 170.10, 169.73, 169.71 (m, 8 × OCOCH3), 98.23 (C-1A), 95.17 (C-1B), 81.01 (C-12′A), 78.38 (C-12′B), 70.98 (C-2), 70.25 (C-3), 68.80 (C-4A), 68.63 (C-4B), 67.94 (C-5), 62.21 (C-6A), 62.08 (C-6B), 59.86 (C-9′A), 59.14 (C-9′B), 56.69 (C-10′A), 54.32 (C-10′B), 51.61 (–OCH3), 40.53, 35.67, 35.37, 34.04, 33.70, 31.75, 29.50, 29.24, 29.00, 28.66, 28.63, 27.69, 25.08, 24.87, 22.62, 22.56, 21.00, 20.82, 20.71, 20.63 (m, 2 × (CH2)13), 14.21 (C-18′A); 14.17 (C-18′B); HR MS m/z calculated for C35H54Br2O12 [M + Na]+ = 823.1880, found 823.1880, 825.1863 [M + 2 + Na]+.
Methyl 9,10-dibromo-12-O-(β-D-glucopyranosyl)-ricinoleate (48). The title compound was prepared from 47 (45 mg, 0.06 mmol) following the general procedure described above. The reaction time was 0.5 h; and 48 (31 mg, 87%) was obtained as a thick syrup. [α]23D = +37.1° (c = 0.75, CHCl3); 1H NMR (400 MHz, CD3OD) (the two enantiomers are marked A & B) δ, 4.89–4.78 (2 × d, J1,2 = 4.0 Hz, 2H, H-1A, H-1B), 4.39–4.35 (m, 2H, H-4A, H-4B), 4.26–4.22 (m, 2H, H-3A, H-3B), 4.14–4.10 (m, 2H, H-5A, H-5B), 3.81–3.48 (m, 16H, 2 × H-12′, 2 × H-6a, 2 × H-6b, 2 × –OCH3, 2 × H-9′, 2 × H-10′), 3.30–3.24 (m, 2H, H-2A, H-2B), 2.24–1.22 (m, 52H, 2 × –(CH2)13), 0.88 (t, J18′,17′ = 6.9 Hz, 6H, 2 × CH3); 13C NMR (100 MHz, CD3OD) δ 174.66 (2 × C-1′), 101.14 (C-1A), 98.61 (C-1B), 78.72 (C-12′A), 76.99 (C-12′B), 73.59, 73.52, 72.69, 72.47, 72.42, 72.13 (m, 2 × C-6, 2 × C-9′, 2 × C-10′), 70.36 (C-2A), 70.22 (C-2B), 61.15, 61.03, 60.92, 59.58, 57.42, 55.43, 53.32 (m, 2 × C-5, 2 × C-3, 2 × C-4) 50.61 (2 × OCH3), 43.26, 40.88, 37.34, 36.55, 35.53, 34.23, 33.44, 31.54, 29.15, 29.01, 28.71, 28.68, 28.65, 28.41, 28.39, 27.18, 27.07, 24.84, 24.71, 24.56, 22.30, 22.24 (m, 2 × (CH2)13), 13.01 (C-18′A); 12.99 (C-18′B); HRMS m/z calculated for C25H46Br2 O8 [M + Na]+ = 655.1457, found 657.1448 [M + 2 + Na]+.
Methyl 12-O-benzyl-ricinoleate (49). NaH (46 mg, 0.96 mmol) was added to a solution of methyl ricinoleate (7, 200 mg, 0.6 mmol) in DMF (5 mL) at 0 °C and the mixture was stirred for 15 min. BnBr (0.1 mL, 0.76 mmol) was then added to the mixture and the temperature was allowed to be brought to 10 °C, and the stirring was continued for 10 h. At this time TLC showed complete disappearance of 7. MeOH (1 mL) followed by triethyl amine (1 mL) were added to the mixture. After stirring for a few minutes the reaction mixture was diluted with diethyl ether and was washed successively with water (×3) and brine in a separatory funnel in the cold. The ether layer was then dried over anhydrous Na2SO4 and was concentrated to dryness under reduced pressure. Purification of the product by chromatography on silica (eluent, EtOAc
:hexane) yielded 49 and 49a (140 mg; 1:0.5 by 1H NMR) as a mixture. The mixture (30 mg) was therefore dissolved in anhydrous MeOH and was treated with NaOMe (catalytic) for 2 h at room temperature. Amberlite IR 120 H+ resin was then added to it to acidic pH and the solution on concentration to dryness and filtration through a short column of silica afforded the title compound neat (20 mg, 72%). [α]23D = +19.3° (c = 0.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.37–7.22 (m, 5H, Ph-CH), 5.48–5.35 (m, 2H, H-9, H-10), 4.57 (d, JA,B = 11.7 Hz, 1H, Ph-CHH), 4.49 (d, JA,B = 11.7 Hz, 1H, Ph-CHH), 3.66 (s, 3H, –COOCH3), 3.40 (p, J = 6.0 Hz, 1H, H-12), 2.36–2.23 (m, 4H, 2 × H-2, 2 × H-11), 2.03 (q, 2H, J = 6.4 Hz, J = 6.7 Hz, 2 × H-8), 1.65–1.22 (m, 20H –(CH2)10), 0.88 (t, J18,17 = 6.8 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.43 (C-1), 139.20 (Ph-C), 131.85 (C-9), 128.40, 127.86, 127.52 (Ph-C), 125.69 (C-10), 79.14 (C-12), 71.07 (Ph-CH2), 51.57 (–OCH3), 34.25 (C-2), 34.13 (C-13), 32.02 (C-11), 31.80, 29.71, 29.59, 29.32, 29.29, 29.27, 27.57, 25.59, 25.10, 22.79 (m, –(CH2)9–), 14.09 (C-18); HR MS m/z calculated for C26H42 O3 [M + Na]+ = 425.3032, found 425.3035.
Methyl 12-O-methyl-ricinoleate (50). The title compound was prepared from 7 (313 mg, 1 mmol) using NaH (70 mg, 1.5 mmol) and MeI (0.09 mL, 1.5 mmol) in DMF (5.0 mL) by the same procedure as described for 49. The reaction time was 4 h and product obtained (210 mg) was a mixture (1
:0.5 by 1H NMR) of the ester 50 and the lactone 50a. Treatment of the mixture (60 mg) with a solution of anhydrous HCl in MeOH (prepared by reacting 0.02 mL thionyl chloride with anhydrous MeOH, 3 mL, at −5 °C) under the nitrogen atmosphere for 8 h at room temperature, followed by concentration of the reaction mixture to dryness and purification of the residue by filtration through a column of silica (eluent, EtOAc:hexanes) gave 50 as a neat product (32 mg, 80% based on 50 weight). [α]23D = +17.2° (c = 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.46–5.33 (m, 2H, H-9, H-10) 3.65 (s, 3H, –COOCH3), 3.32 (s, 3H, –OCH3), 3.17 (p, J = 5.8 Hz, 1H, H-12), 2.31–2.15 (m, 4H, 2 × H-2, 2 × H-11), 2.01 (q, 2H, J = 6.5 Hz, J = 6.7 Hz, 2 × H-8), 1.65–1.53 (m, 2H, 2 × H-7), 1.48–1.37 (m, 2H, 2 × H-13), 1.37–1.21 (m, 16H –(CH2)8–), 0.86 (t, J18,17 = 6.6 Hz, 3H, –CH3); 13C NMR (100 MHz, CDCl3) δ 174.23 (C-1), 131.67 (C-9) 125.42 (C-10), 80.97 (C-12), 56.54 (–OCH3), 51.39 (–COOCH3), 34.07 (C-2), 33.57 (C-13), 31.86 (C-11), 31.86, 31.06, 29.54, 29.47, 29.11, 27.38, 25.34, 24.93, 22.61, (m, –(CH2)9–), 14.06 (C-18); HR MS m/z calculated for C20H38O3 [M + Na]+ = 349.2719, found 349.2714.
Conclusions
In conclusion, 28 novel glycosides of ricinoleic acid were synthesized and their antimicrobial activity was evaluated, seven of them showing promising wide spectrum antibacterial activity against Gram +ve bacteria. Two compounds, 29 (NP-2672) and 39 (NP-2689), showed good to excellent activity against various non-clinical/clinical/NorA overexpressed/resistant strains of S. aureus as well as other Gram +ve bacteria. These results suggest that carbohydrate moiety in the glycosides of the ricinoleic acid played an important role for imparting the antibacterial activity by increasing the membrane permeability. It is presumably because of binding of these molecules with D-Ala-D-Ala, preventing the synthesis of the long polymers of N-acetylmuramic acid and N-acetylglucosamine that form the backbone strands of the bacterial cell wall. In this sense, these compounds could be potential candidates for therapeutics as they may not be targeting the cell components such as nucleic acids and proteins. The future studies in this context shall therefore be for answering these questions.Author contributions
All authors have given approval to the final version of the manuscript.Acknowledgements
K. R. K. sincerely acknowledges financial support in the form of a Senior Research Fellowship by CSIR, India. H. N. and R. T. sincerely acknowledge financial support from CSIR-Network Project (BSC-211) to carry out the work and the Department of Biotechnology, Govt. of India for Junior Research Fellowship, respectively. This work is dedicated to Chandravathi Kuppala and Gangaraju Kuppala.Notes and references
- S. Rachakonda and L. Cartee, Curr. Med. Chem., 2004, 11, 775–793 CrossRef CAS PubMed
.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010–2023 CrossRef CAS
- A. Raja, J. LaBonte, J. Lebbos and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 943–944 CrossRef CAS
- J. D. Alder, Drugs Today, 2005, 42, 81–90 CrossRef PubMed
- R. M. Klevens, M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. Mcdougal, R. B. Carey and S. K. Fridkin, Invasive Methicillin-Resistant Staphylococcus aureus Infect, United States, 2007, vol. 298, pp. 1763–1771 Search PubMed
- M. S. Butler, K. A. Hansford, M. A. Blaskovich, R. Halai and M. A. Cooper, J. Antibiot., 2014, 67, 631–644 CrossRef CAS PubMed
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483–490 CrossRef CAS
- M. S. Butler and M. A. Cooper, J. Antibiot., 2011, 64, 413–425 CrossRef CAS
- Y. Zhang, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 529–564 CrossRef CAS PubMed
- D. Chatterjee, Curr. Opin. Chem. Biol., 1997, 1, 579–588 CrossRef CAS PubMed
- C. J. Zheng, J.-S. Yoo, T.-G. Lee, H.-Y. Cho, Y.-H. Kim and W.-G. Kim, FEBS Lett., 2005, 579, 5157–5162 CrossRef CAS PubMed
- M. Niwano, E. Mirumachi, M. Uramoto and K. Isono, Agric. Biol. Chem., 1984, 48, 1359 CrossRef CAS
- W. P. Larson, W. F. Cantwell and T. B. Hartzell, J. Infect. Dis., 1919, 25, 41–47 CrossRef CAS
- B. Y. Charles and G. Lewis, Biochem. J., 1923, 17, 813–816 CrossRef
- C. P. Miller and R. Castles, J. Bacteriol., 1931, 22, 339–348 CAS
- S. F. Seeley, Ann. Surg., 1932, 96, 350–358 CrossRef CAS PubMed
- A. F. Novak, G. C. Clark and H. P. Dupuy, J. Am. Oil Chem. Soc., 1961, 38, 321–324 CrossRef CAS
- F. A. Andersen, Final report on the safety assessment of Ricinus communis (castor) seed oil, hydrogenated castor oil, glyceryl ricinoleate, glyceryl ricinoleate se, ricinoleic acid, potassium ricinoleate, sodium ricinoleate, zinc ricinoleate, cetyl ricinoleate, ethyl ricinoleate, glycol ricinoleate, isopropyl ricinoleate, methyl ricinoleate, and octyldodecyl ricinoleate, Int. J. Toxicol., 2007, 26(3), 31–77 Search PubMed.
- Y. T. Osaka and S. K. Osaka, US 2010/0221197 A1, 2010
- C. D. R. M. D'Oca, T. Coelho, T. G. Marinho, C. R. L. Hack, R. da Costa Duarte, P. A. da Silva and M. G. M. D'Oca, Bioorg. Med. Chem. Lett., 2010, 20, 5255–5257 CrossRef PubMed
- Y. Mohini, R. B. N. Prasad, M. S. L. Karuna, Y. Poornachandra and C. Ganesh Kumar, Bioorg. Med. Chem. Lett., 2014, 24, 5224–5227 CrossRef CAS PubMed
- G. Borsotti, G. Guglielmetti, S. Spera and E. Battistel, Tetrahedron, 2001, 57, 10219–10227 CrossRef CAS
- A. Bouzide and G. Sauvé, Tetrahedron Lett., 1997, 38, 5945–5948 CrossRef CAS
- E. E. Dueno, F. Chu and K. W. Jmlg, Tetrahedron Lett., 1999, 40, 1843–1846 CrossRef CAS
- G. Westphal, B. Reukauf, S. Storm and A. Bartels, Food Nahrung, 1994, 38, 78–81 CrossRef CAS
- Y. V. Tomilov, M. S. Yunusov and O. M. Nefedov, Russ. J. Org. Chem., 2001, 37, 608–611 CrossRef
- A. Smith, P. Nobmann, G. Henehan, P. Bourke and J. Dunne, Carbohydr. Res., 2008, 343, 2557–2566 CrossRef CAS PubMed
- H. Lu and P. J. Tonge, Acc. Chem. Res., 2008, 41, 11 CrossRef CAS PubMed
- J. Schiebel, A. Chang, H. Lu, M. V. Baxter, P. J. Tonge and C. Kisker, Structure, 2012, 20, 802 CrossRef CAS PubMed
- L. Chopra, G. Singh, K. Kumar Jena and D. K. Sahoo, Sci. Rep., 2015, 5, 13412 CrossRef CAS PubMed
- G. W. Kaatz and S. M. Seo, Antimicrob. Agents Chemother., 1997, 41, 2733–2737 CAS
- M. Stavri, R. Schneider, G. O'Donnell, D. Lechner, F. Bucar and S. Gibbons, Phytother. Res., 2004, 18, 774–776 CrossRef CAS PubMed
- R. F. Epand, J. E. Pollard, J. O. Wright, P. B. Savage and R. M. Epand, Antimicrob. Agents Chemother., 2010, 54, 3708–3713 CrossRef CAS PubMed
- C. L. Nobles, S. I. Green and A. W. Maresso, PLoS Pathog., 2013, 9, e1003507 CAS
- N. K. Patel and K. K. Bhutani, Phytomedicine, 2014, 21, 946–953 CrossRef CAS
- K. P. R. Kartha and H. J. Jennings, J. Carbohydr. Chem., 1990, 9, 777–781 CrossRef CAS
- G. Mugunthan, K. Ramakrishna and K. P. R. Kartha, Trends Carbohydr. Res., 2010, 2, 20–27 CAS
- R. K. Ness, H. G. Fletcher and C. S. Hudson, J. Am. Chem. Soc., 1951, 73, 296–300 CrossRef CAS
- S. Sinha, R. Srivastava, E. de Clercq and R. K. Singh, Nucleosides, Nucleotides Nucleic Acids, 2004, 23, 1815–1824 CAS
- G. J. F. Chittenden, Carbohydr. Res., 1972, 25, 35–41 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20136e |
| This journal is © The Royal Society of Chemistry 2016 |