
Determination of losartan potassium in the presence of hydrochlorothiazide via a combination of magnetic solid phase extraction and fluorometry techniques in urine samples
Abstract
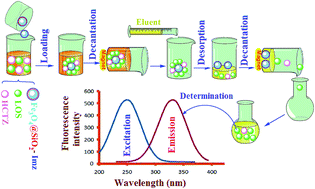
A sensitive and highly efficient method for the determination of losartan potassium (LOS) in the presence of hydrochlorothiazide (HCTZ) via a combination of magnetic solid phase extraction (MSPE) and fluorometry techniques is suggested. For this purpose, imidazolium ionic liquid (Imz)-modified Fe3O4@SiO2 nanoparticles (Fe3O4@SiO2-Imz) were utilized as an adsorbent for the MSPE. Fe3O4@SiO2-Imz exhibited excellent extraction performance for LOS, in part due to the anion-exchange groups in the Imz moiety. The experimental parameters, such as sample pH values, the amount of adsorbent, the extraction time, the desorption solvent, and the desorption time, that could affect the extraction performance were examined and optimized. The determination of LOS in the presence of HCTZ in urine samples was carried out using selective and sensitive excitation–emission fluorescence spectroscopy (EEFS), because only LOS exhibited fluorescence emission at 325 nm, while the HCTZ did not exhibit any fluorescence emission. The limit of detection (LOD) for LOS using this technique was 0.12 ng ml−1.
 Please wait while we load your content...
Please wait while we load your content...

