
Hydrogen peroxide treated graphene as an effective nanosheet filler for separation application
Dharupaneedi P. Suhasab,
Tejraj M. Aminabhavic,
Han M. Jeongd and
Anjanapura V. Raghu*e
aMaterials Science Division, Poornaprajna Institute of Scientific Research, Bengaluru 560110, India
bDepartment of Chemistry, St. Joseph’s College, 36, Langford Road, Bengaluru – 560 027, India
cSoniya College of Pharmacy, S.R. Nagar, Dharwad – 580 002, India
dDepartment of Chemistry, University of Ulsan, Ulsan, Republic of Korea 680 749
eCentre for Emerging Technologies, Jain Global Campus, Jain University, Ramanagaram – 562 112, India. E-mail: gsraghu2003@yahoo.co.in
First published on 9th November 2015
Abstract
Graphene has been regarded as an important platform material to develop membranes for a wide range of applications. With state-of-the-art separation technologies, it is highly desirable to develop efficient membranes for organic dehydration that can reduce recovery costs. In this work, graphene loaded polymer composites of chitosan and poly(vinyl pyrrolidone) are used for pervaporation (PV) applications because the presence of a filler in the polymer matrix would boost membrane performance. Such an enhancement of barrier properties compared to conventional nascent membranes is a result of improved interface compatibility and interaction. In this research, H2O2 treated graphene was used as a nanofiller in a chitosan–poly(vinyl pyrrolidone) blend matrix to develop membranes that were tested for PV dehydration of ethanol as a function of filler loading, feed composition and temperature. Physico-chemical interactions between filler nanoparticles and the polymer matrix are responsible for the improved performance. The prepared membranes have been characterized via several analytical techniques. The 2.5 wt% H2O2 treated graphene loaded composite membrane offered a selectivity of 955, which is almost 40% higher than those membranes loaded with the same quantity of untreated fillers. Such enhanced membrane performance is attributed to an increase in the number of oxygen functionalities on the graphene surface after H2O2 treatment, resulting in improved filler interaction at the interface of the polymer and graphene in the presence of permeate molecules. Calculations involving the Flory–Huggins parameter, diffusion coefficient and Arrhenius activation energy barrier have been performed to explain the PV results in terms of the observed increase in membrane performance.
Introduction
The increase in global demand for renewable fuels as well as the depletion of fossil fuel reserves have lead to a search for alternate energy sources. Among the vast array of chemicals derived from renewable sources, ethanol is a key platform chemical used in a wide range of applications. Ethanol obtained from the fermentation of biomass is most often used as a motor fuel or fuel additive to gasoline.1,2 The production of bio-ethanol essentially involves hydrolysis, fermentation and purification. As per the European Union, fuel grade ethanol should contain at least 98.7 wt% of ethanol, which makes the purification step crucial.3During purification, 10 wt% of ethanol (obtained from fermentation) is concentrated up to 99 wt%, which is fuel grade. However, distillation is good up to a composition of 80–85 wt% ethanol and above which the method becomes prohibitively expensive, especially near an azeotropic composition4,5 and thus a major share of the production cost goes to alcohol purification. On the other hand, membrane-based pervaporation (PV) offers an alternative approach with the advantages of low energy consumption, low operating cost and excellent performance in dehydrating ethanol at azeotropic compositions.6 The method is vital to enhance the ethanol composition from 80–85 wt% up to 99 wt%, but key to this success relies on developing suitable membranes that can offer simultaneous high flux and selectivity along with a good thermo-mechanical strength.7,8
Hydrophilic polymers such as sodium alginate, chitosan (CS), poly(vinyl alcohol), polyacrylonitrile and polyimide have been widely used in the earlier literature.9 Among these CS, a biopolymer obtained from crustacean shells which has good film forming ability, moderate mechanical strength as well as an excellent affinity towards water, is regarded as one of the indispensable materials for PV dehydration of organics.10 Poly(vinyl pyrrolidone) (PVP) is another hydrophilic biopolymer having excellent compatibility with CS to form a homogeneous blend matrix.11 Earlier research reported that blending of PVP with CS can influence the PV performance of membranes.12,13
It has been realized that the performance of chitosan for extended use can be reduced due to its swelling characteristics. To avoid this issue, various methods such as blending, crosslinking etc., have been attempted. Even compared to such membranes, recent trends for developing nanocomposite membranes prepared by incorporating nanofillers into a polymer matrix seem to offer improved physical, chemical and separation properties. Such nanocomposite membranes combine the flexibility of polymers with the strength and specificity of inorganic fillers.14 Typical fillers used in developing nanocomposite membranes are usually hydrophilic, porous or intercalated materials, whose presence creates tortuous pathways in a matrix, thus favouring selective permeation of smaller molecules such as water.15 Some of the widely used fillers in this category are clay,16,17 heteropolyacids,18 zeolites,19 single-walled alumina-silicate nanotubes,20 zeolitic imidazolate frameworks (ZIFs),21 titanate nanotubes,22 carbon nanotubes23 and graphene.24
Graphene is a two dimensional crystalline allotrope of carbon with densely packed carbon atoms arranged in a regular hexagonal sp2 bonded atomic scale chicken wire pattern. In recent years, graphene has created greater interest as a versatile nanosheet material due to its exceptional electron transport capacity, superior mechanical strength and high surface area.25 These properties have prompted researchers to develop nanocomposite polymeric membranes containing graphene.26–32 Earlier, Nair33 et al. and Hung34 et al. used submicron thick membranes of graphene oxide (GO) for separating various solvents, which showed superior selectivity to water compared to organics. Unfortunately, practical applications of these membranes are limited due to the high cost involved in graphene production. An effective approach would therefore be to use graphene as a filler in a polymer matrix to develop graphene-loaded nanocomposite membranes. However, uniform dispersion of graphene in a polymer matrix is somewhat difficult, yet previous efforts have been fruitful.24,35 Basically, the presence of hydroxyl, carboxyl and epoxide groups on the graphene surface would achieve electrostatic and H-bonded interactions with the polymer matrix. It is thought that if we use oxygen containing functional groups on the graphene surface, it would enhance interfacial interaction between the polymer structure and nanosheets of graphene, consequently resulting in enhanced membrane performance.
In efforts to enhance interface interactions between graphene and the chitosan polymer, we have treated graphene with hydrogen peroxide, which introduces additional oxygen functionalities on the graphene surface. While using these nanofillers to prepare nanocomposite blend membranes of chitosan/PVP, the presence of PVP is expected to facilitate filler dispersivity. The prepared nanocomposite membranes were tested for PV dehydration of ethanol. The interaction between graphene oxide and the polymer matrix was assessed via Fourier transform infrared spectroscopy (FTIR), field emission scanning electron microscopy (FE-SEM), wide angle X-ray scattering (WAXS) and contact angle measurements. Furthermore, membrane performance was evaluated by sorption, diffusion and Arrhenius activation energy parameters.
Experimental
Materials and methods
Natural graphite (HC-908) was procured from Hyundai Coma Co. Ltd., Mumbai, India. Chitosan and poly(vinyl pyrrolidone) polymers were purchased from SRL chemicals, Mumbai, India. Glacial acetic acid (100%), hydrogen peroxide, concentrated HCl (35%), nitric acid, potassium chloride and glutaraldehyde were all procured from S.D. Fine Chemicals, Mumbai, India. Absolute ethanol (EtOH) (99.9%) was obtained from Commercial Alcohols, Brampton, Canada. All other chemicals used in this work were of reagent grade and used without further purification (unless specified). Double-distilled water was used throughout the study.Preparation of functionalized graphene sheets
Graphite oxide was prepared using a modified Brondie method as reported by Jeong36 et al. In a typical procedure, 10 g of nascent graphite was placed in a 500 mL round bottom flask to which 200 mL of ice cooled fuming nitric acid was added. Subsequently, the reactants were mixed overnight and transferred to a beaker containing 3 L of double distilled water. The product thus obtained was filtered and washed with copious amount of water for neutralization. Graphite oxide has the empirical formula: C10O3.45H1.58 (as per elemental analysis). Dried graphite oxide was introduced into a quartz tube maintained under an inert atmosphere. The quartz tube was introduced into a furnace maintained at 1100 °C, where layers of graphite oxide disintegrate into individual graphene sheets. Thus prepared graphene has the empirical formula: C10O0.78H0.38 with a surface area of 428 m2 g−1.H2O2 treatment of graphene sheets
In order to enhance oxygen functionality, graphene was immersed in an excess amount of H2O2 solution (30 wt% concentration) and the mixture was sonicated for 1 h followed by stirring for 3 h at 60 °C. The products were filtered and washed with excess amount of double distilled water followed by acetone washing and dried at 100 °C in a hot air oven for 24 h. Thus obtained graphene showed an enhanced oxygen content as per the empirical formula C10O0.99H0.73 and is designated as graphene oxide (GO).36 The enhanced oxygen functionality is expected to increase the compatibility of graphene with the chitosan matrix. The morphology of GO was examined by TEM, XRD, FTIR and BET studies.Membrane fabrication
All the blend membranes containing 10 wt% of PVP in CS and loaded with GO as a nanofiller were prepared by solution casting followed by solvent evaporation. First, a chitosan solution was prepared by mixing chitosan powder with acetic acid and water in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:94. Then, a PVP solution prepared in water media was added to the above mixture, stirred on a magnetic stirrer and allowed to stand overnight to get rid of air bubbles. The required quantity of GO (1, 2, 2.5 and 3 wt%) was agitated and sonicated for 2 h with water before adding into the CS–PVP blend solution that was stirred on a magnetic stirrer for 24 h to achieve uniform dispersion. The resulting mixture was poured onto a perfectly aligned clean glass plate kept in a dust free environment and allowed to dry at the ambient temperature.
:3:94. Then, a PVP solution prepared in water media was added to the above mixture, stirred on a magnetic stirrer and allowed to stand overnight to get rid of air bubbles. The required quantity of GO (1, 2, 2.5 and 3 wt%) was agitated and sonicated for 2 h with water before adding into the CS–PVP blend solution that was stirred on a magnetic stirrer for 24 h to achieve uniform dispersion. The resulting mixture was poured onto a perfectly aligned clean glass plate kept in a dust free environment and allowed to dry at the ambient temperature.
Dried membranes were immersed in a cross-linking bath for 2 h containing a water–acetone mixture in a ratio 3:7 along with 2 mL of glutaraldehyde as a cross-linking agent and 1 mL of HCl as a catalyst. In order to remove the unreacted glutaraldehyde, membranes were alternately rinsed with water and methanol about 4–5 times followed by soaking in methanol for 24 h. Using the above procedure 1, 2, 2.5 and 3 wt% of GO loaded CS–PVP blend nanocomposite membranes were prepared and designated as CS–PVP-1, CS–PVP-2, CS–PVP-2.5 and CS–PVP-3, respectively. Nascent CS (crosslinked, but without adding a filler) and CS–PVP-2.5 (U) (CS–PVP by adding 2.5 wt% of untreated graphene) were fabricated as control membranes for comparison purposes.
Membrane characterization
The chemical interactions between functional groups of GO and the CS–PVP blend matrix were assessed using FTIR. All measurements were done in transmission mode in the range of 400–4000 cm−1 using a Bruker Alpha-T spectrophotometer. Each sample was scanned 32 times with a resolution of 4 cm−1. Prior to this, the sample pellets were prepared by applying a hydraulic pressure of 400–450 kg cm−2 on a mixture containing dried KBr and the sample.Solid state morphology of the membranes was analyzed using a Bruker D-2 phaser X-ray diffractometer. The Cu-Kα radiation source was used to generate X-rays of 1.5406 Å wavelength. The powder samples were scanned in the range of 2θ of 5–60° at a scanning rate of 2° min−1.
The micro-morphology and thickness of the nanocomposite membranes were assessed using a FE-SEM (Field Emission Scanning Electron Microscopy) Zeiss Ultra-55 instrument. In order to enhance the image quality, membranes were coated with a conductive layer of sputtered gold of 0.5–1 μm thickness.
Thermal stability and degradation patterns of all the membranes were evaluated by thermogravimetric analysis (TQ-500) performed in the temperature range of 40–600 °C under a slow heating rate of 10 °C min−1 in an inert atmosphere by flowing nitrogen gas at a constant rate of 50 mL min−1. For each analysis, approx. 5–8 mg of the sample was put in an aluminium pan and thermal stability was analyzed.
A Mettler Toledo (DSC-822) instrument was used to obtain DSC (differential scanning calorimetry) thermograms of the membranes. The aluminum pans were conditioned before the start of the experiment. About 10 mg of the sample was sealed in an aluminum pan and measurements were taken from ambient temperature to 250 °C with a ramp rate of 10 °C min−1 while maintaining an inert atmosphere through the constant supply of nitrogen gas at a flow rate of 50 mL min−1.
The relative surface hydrophilicity of the membranes was measured by a static water sessile-drop contact angle instrument (Data Physics OCA-20) at 25 °C. Membrane samples were vacuum dried prior to obtaining contact angle measurements. Membranes were adhered to a suitable glass surface, onto which deionized water droplets of 2 μL were placed and their image was photographed at exactly 10 s using a CCD camera. All the reported values are averages of five measurements taken at different places by considering standard deviations of ±3%.
Equilibrium swelling experiments were performed gravimetrically at 30 °C by soaking circularly-cut membrane samples (2.5 cm dia) in 10 wt% of water containing a feed mixture. The initial weight of the membrane was noted as Wd. These membranes were immersed in air tight test bottles containing 30 cm3 of the feed mixtures for 48 h. Then, membranes were removed from the test bottles, wiped-off with soft tissue paper wraps to remove the surface-adhered liquid droplets and the swollen weight was noted as Ws. From these data, % equilibrium swelling was calculated as:37
 | (1) |
Pervaporation experiments
An indigenously designed stainless steel unit was used to carry out the PV experiments.38 The set-up had two compartments viz., a feed tank and permeate cell. The membrane with an effective area of 3.84 × 10−3 m2 was placed tightly between two O-shaped Teflon rings, which was further placed onto a porous stainless steel support. The feed tank was a double-walled cylindrical shaped stainless steel piece of equipment with a maximum capacity of 500 mL. Through its outer jacket, warm water from a thermostatic bath (Grant UK Model GD-120) was circulated to maintain the desired temperature in the feed tank, which was provided with a stirrer and a thermometer; the feed mixture was maintained at atmospheric pressure (760 mmHg), while the permeate sample was maintained at >5 mbar pressure with the help of a Telstar double stage suction pump. To attain constant flux, membranes were equilibrated with the feed mixture for 2 h prior to starting the experiment. Permeate samples were collected at regular intervals of time using two glass traps immersed in a liquid nitrogen containing Dewar flask.Samples were collected at intervals of 1 h and their weight was measured on a Sartorius BSA 224 balance, while compositions of the feed and permeate samples were measured by a refractometer as well as using gas chromatography (GC). The refractive index was measured using a Mettler Toledo Refractometer to an accuracy of ±0.0001 units and the % composition of the permeate mixture was estimated with the help of the previously established calibration curve of the refractive index vs. known composition of the mixture at 30 ± 0.1 °C. To reconfirm the permeate compositions, GC runs were made on a Thermofisher Trace-700 coupled with a Porapack Q column and TCD detector. In this, 1 μL of the sample was injected along with nitrogen carrier gas at a flow rate of 1.5 μL min−1 at an oven temperature of 150 °C. For each sample, three separate readings were taken but the average values of three independent measurements were considered to an accuracy of ±3% of the standard error. Membranes were stable for more than 10 h of continuous PV operation.
As recently pointed out,39 normalizing the flux with respect to the driving force would make the comparison of PV data more meaningful. It may significantly decouple the effect of the operating conditions on performance evaluation, while at the same time quantify the contribution by the nature of the membrane separation efficiency. In this work, as noted previously,40,41 the driving force normalized parameters viz., the permeability (PGi) and selectivity (αij) values proposed by Baker and Wijmans,42 are used to treat the PV data as these appear to be more appropriate to represent the PV results in a more meaningful manner. Moreover, such data can be comparable directly with the literature findings. This prompted us to compute permeability (PGi) using:
 | (2) |
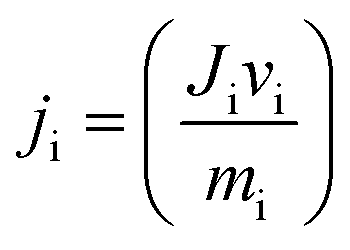 | (3) |
The partial vapor pressure (pfi) was calculated using the van Laar equation:
| pfi = xiγipsi | (4) |
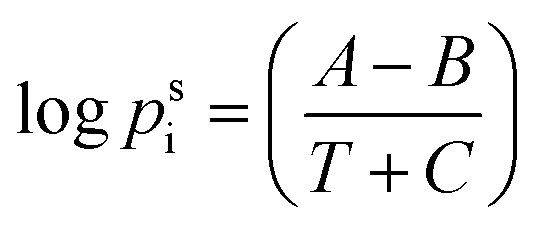 | (5) |
 | (6) |
Results and discussion
Characterization of graphene oxide (GO)
TEM images of GO shown in Fig. 1a and b reveal a dark thin crumpled paper-like surface with many fibrous types of wrinkles and foldings, which are characteristics of graphene sheets.45 In order to examine the % composition of oxygen on the graphene surface, SEM-EDX analysis was performed as shown in Fig. 1c and d, wherein green dots on the surface are attributed to the presence of oxygen, revealing a highly oxygenated surface. The presence of oxygen containing functional groups is also evident from the examination of the FTIR spectrum given in Fig. 1e, where absorption bands are observed at 3430, 1540 and 1230 cm−1 related to the oxygen containing functional groups of GO. | ||
| Fig. 1 (a) TEM image of GO; (b) (inset) TEM image at higher resolution; (c) EDX-image of GO; (d) EDX analysis showing the weight% composition of GO; (e) FTIR spectrum of GO; (f) XRD of GO. | ||
The 2D (0 0 2) sheet like structure of graphene was marked with the appearance of a XRD peak at 2θ of 26.3° (as seen in Fig. 1f) showing a d-spacing of 0.339 nm. The surface area of graphene as measured by the BET isotherm was 428 m2 g−1, revealing a six-layered structure. The elemental analysis confirms that after H2O2 treatment, oxygen content increased from C10O0.78H0.38 to C10O0.99H0.73. All these unique observations are attributes of graphene, which are expected to enhance the overall performance of the nanocomposite membranes in PV dehydration of ethanol.
Characterization of membranes
Fourier transform infrared spectroscopy (FTIR)
FTIR spectra of nascent CS–PVP and its GO incorporated nanocomposite membranes are presented in Fig. 2, wherein all the spectra are shifted upwards to avoid overlapping. The FTIR spectrum of nascent CS–PVP shows multiple absorption bands in the region of 400–4000 cm−1, which are in accordance with the reported spectra by Magalad18 et al. The combined characteristic broad peak of the –OH and –NH2 groups was observed around 3400–3500 cm−1. Interestingly, these peaks show a red shift with increasing filler loading, signifying an effective hydrophilic linkage between the filler particles and the polymer blend matrix.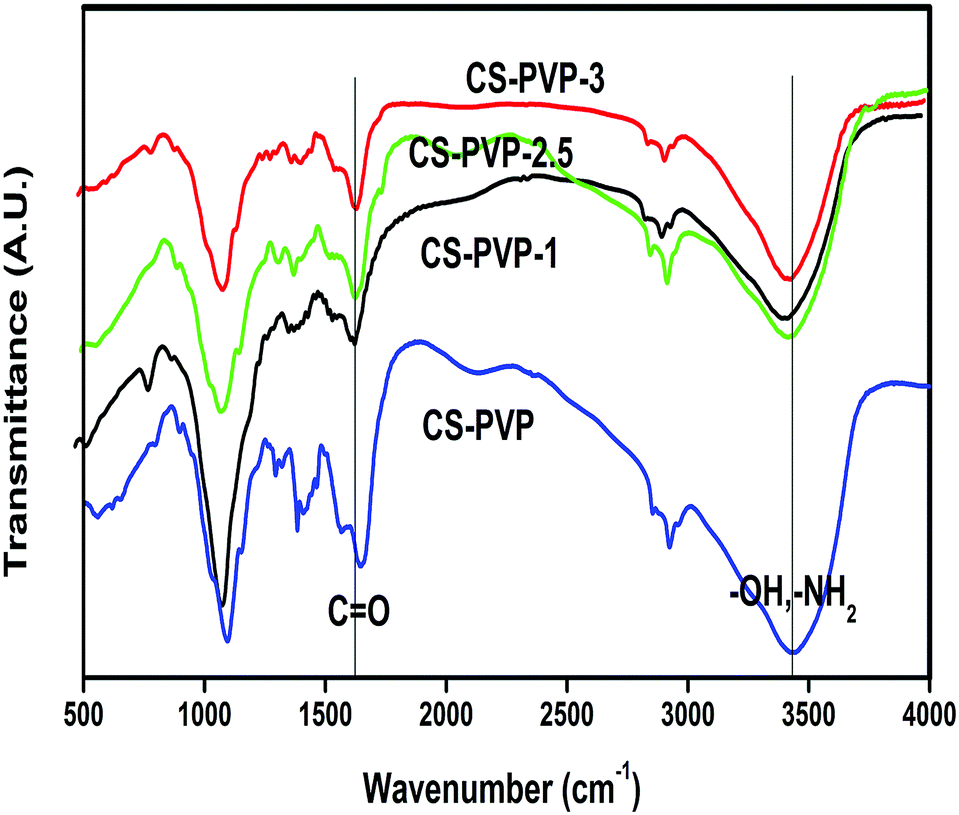 | ||
| Fig. 2 FTIR of nascent CS–PVP and its composites with different loading of GO, showing an increase in the red-shift with increasing GO loading. | ||
Wide angle X-ray scattering (WAXS)
Fig. 3 depicts WAXS tracings of nascent CS–PVP and its nanocomposites, showing two characteristic peaks. The first sharp peak observed at 2θ of 10–12°, and the second weak peak observed at 18–22°, are in agreement with the reported data.13 With an increase in GO loading, the peak intensity gradually decreased due to molecular level interactions between GO and CS/PVP, leading to a disturbance in the ordered arrangement of the CS chains, consequently resulting in a decrease of peak intensity. This observation indicates fine molecular level dispersion of the GO nanosheets in the CS/PVP matrix and this might have facilitated the tortuous pathways in the matrix, thereby favouring selective diffusion of water molecules over that of ethanol. This can be further confirmed by the non-appearance of the GO peak. | ||
| Fig. 3 XRD of nascent CS–PVP and its composite membranes with different loadings of GO, showing a decrease in peak intensity with increasing GO loading. | ||
Field emission scanning electron microscopy (FE-SEM)
To observe the dispersion of the GO nanosheets at higher resolution and to obtain additional information on the interfacial interaction between GO and the polymer matrix, SEM images were taken. The cross-sectional view of the nascent CS/PVP blend is shown in Fig. 4(a), which appears much darker due to the non-conductive nature of CS/PVP towards electrons. Obviously, brighter spots observed in the GO loaded CS/PVP blend-composite membranes as shown in Fig. 4(b–g) are attributed to the presence of the GO sheets. Both the cross-sectional and top views of the nanocomposite membranes were obtained. Fig. 4(e–g) shows cross-sectional SEM images of CS–PVP-1, CS–PVP-2.5 and CS–PVP-3, respectively. These images are densely packed with groves due to differential interactions between GO and the CS–PVP matrix. | ||
| Fig. 4 FE-SEM (top-view) of (b) CS–PVP-1, (c) CS–PVP-2.5, and (d) CS–PVP-3; FE-SEM (cross-sectional view) of (a) nascent CS–PVP, (e) CS–PVP-1, (f) CS–PVP-2.5 and (g) CS–PVP-3; insets show SEM images at higher resolution. | ||
The top surface of all the GO incorporated CS/PVP membranes are shown in Fig. 4(b–d). These images show a rougher surface as compared to the smoother surface observed for the nascent CS/PVP membrane. Notice that the presence of bright spots increases with GO loading. The higher resolution images as shown in the insets of Fig. 4(b–d) reveal individual particle dispersion of CS–PVP-1 and CS–PVP-2.5 membranes, but the CS–PVP-3 membrane showed micro-agglomeration. These observations suggest that membranes seem to be reaching an optimal performance limit at around 2.5 wt% loading, and above this loading limit, particles start agglomerating, thus resulting in a reduced performance. Since the GO nanosheets are well dispersed in the membrane blend matrix, and hardly any individual nanosheets could be seen, this confirms the superior interfacial interaction. The average membrane thickness was estimated to be around 82 μm.
Thermogravimetric analysis (TGA)
Thermal analyses of all the membranes were performed in the temperature range of 40–600 °C in an inert nitrogen atmosphere, and the results displayed in Fig. 5a show a combination of a release of moisture followed by breaking of the polymer chains. The first major weight loss occurred at 100 °C, due to the release of moisture. The second major weight loss of about 30–40 wt% occurred at 230 °C, which can be attributed to polymer chain degradation. Thus by increasing GO loading, thermal stability improved, indicating a favourable interface interaction between GO and the polymer blend matrix. | ||
| Fig. 5 Thermal analysis of nascent CS–PVP, CS–PVP-1, CS–PVP-2.5 and CS–PVP-3 using (a) non-oxidative TGA curves and (b) DSC thermograms. | ||
Differential scanning calorimetry (DSC)
DSC analyses of all membranes were performed from 40 to 350 °C under a nitrogen (inert) atmosphere and thermograms are presented in Fig. 5b. An endothermic curve is observed from 96 to 130 °C, whereas an exothermic curve in the temperature range of 255 to 275 °C is observed. The endothermic curve is mainly due to the release of absorbed water molecules, while the exothermic curve is due to polymer chain degradation at higher temperature. With increasing GO loading, both endothermic and exothermic curves shifted to higher temperatures, indicating enhanced water holding capacity and thermal stability of the membranes.Contact angle studies
The water contact angle is typically used for estimating the relative hydrophilicity of membranes. Membranes with smaller contact angle values are more hydrophilic and vice versa. The effect of GO loading on membrane hydrophilicity is shown in Fig. 6, where we find a systematic increase in membrane hydrophilicity with an increase in GO loading. This is attributed to enhanced surface hydrophilicity and roughness due to GO loading in the polymer matrix. | ||
| Fig. 6 Change in water contact angle (θ°) of the nanocomposite membranes vs. wt% of GO loading, showing an increase in hydrophilicity with increasing GO loading. | ||
Equilibrium swelling
Fig. 7 presents the % equilibrium swelling (ES) results of nascent CS–PVP as well as other nanocomposite membranes at 30 °C for 10 wt% water containing ethanol feed mixtures. These studies are helpful to analyze free volume (tortuous pathways) of the matrix to solvent diffusion or permeation.46 It is observed that with an increase in filler loading, there is a parallel increase in membrane swelling, which is due to enhanced membrane hydrophilicity. Compared to CS–PVP-2.5 (U), the CS–PVP-2.5 membrane showed higher swelling. This may be due to the fact that H2O2 treatment increases number of oxygen functionalities on the graphene surface, thereby enhancing the number of active sites to facilitate water sorption, but uncontrolled membrane swelling can be avoided or reduced considerably by glutaraldehyde cross-linking. | ||
| Fig. 7 Equilibrium swelling (%) of membranes at 10 wt% of the water–EtOH mixture at 30 °C, showing a decrease in swelling with increasing GO loading. | ||
Membrane performance
Effect of GO loading
Fig. 8 and Table 1 show the effect of GO loading on PV performance of the nanocomposite membranes at 30 °C tested for 10 wt% of water in an ethanol feed mixture. As the GO loading increased from 1 to 2.5 wt%, both permeability and selectivity increased. This could be attributed to the favourable compatibility between GO and CS–PVP, due to uniform dispersion of the GO nanosheets in the blend membrane matrix (as was also shown before by the FE-SEM studies). Contact angle measurements also support that the presence of the GO particles in the membrane interface surface increases the hydrophilic nature, leading to enhanced sorption selectivity. GO helps as a reinforcing bridge in the blend matrix, resulting in tortuous pathways, thus enabling high water selective diffusivity. At a maximum loading of 2.5 wt% of GO, optimum separation performance is observed and above this limit particle agglomeration resulted and at 2.5 wt% GO loading, the maximum selectivity observed is 955. | ||
| Fig. 8 Effect of filler loading on permeability (Barrer) and selectivity of nascent chitosan and GO loaded chitosan nanocomposite membranes at 10 wt% of the water–EtOH feed mixture. | ||
| Membrane type | Feed temp. (°C) | Equilibrium swelling (%) | Permeability (Barrer) | Selectivity (αij) | Dw × 10−13 (m2 s−1) | De × 10−15 (m2 s−1) |
|---|---|---|---|---|---|---|
| Nascent CS–PVP | 30 | 48 | 85 | 152 | 2.9 | 22 |
| 40 | 87 | 135 | 2.8 | 24 | ||
| 50 | 93 | 123 | 3.2 | 30 | ||
| CS–PVP-1 | 30 | 34 | 93 | 294 | 3.1 | 12 |
| 40 | 97 | 236 | 3.5 | 17 | ||
| 50 | 103 | 212 | 3.7 | 20 | ||
| CS–PVP-2 | 30 | 28 | 99 | 624 | 3.2 | 6 |
| 40 | 102 | 472 | 3.7 | 9 | ||
| 50 | 114 | 405 | 4.0 | 11 | ||
| CS–PVP-2.5 | 30 | 22 | 105 | 955 | 3.4 | 4 |
| 40 | 108 | 749 | 3.8 | 6 | ||
| 50 | 118 | 499 | 4.3 | 10 | ||
| CS–PVP-3 | 30 | 20 | 117 | 584 | 3.8 | 7 |
| 40 | 122 | 439 | 4.6 | 12 | ||
| 50 | 128 | 366 | 5.2 | 16 | ||
| CS–PVP-2.5 (U) | 30 | 21 | 101 | 671 | 3.3 | 5 |
| 40 | 104 | 472 | 3.6 | 9 | ||
| 50 | 122 | 407 | 4.2 | 12 |
As observed in Fig. 8, as GO loading increased to 3 wt%, the membrane selectivity value decreased sharply from 955 to 584. This is mainly attributed to increased defects induced by excess GO loading, which would strongly destroy interfacial adhesion between GO and the polymer blend matrix. Even EtOH molecules may also permeate preferentially through such defects and lose interface spaces between GO and the polymer blend matrix, which eventually might have led to an increase in permeability at the cost of selectivity. Moreover, there might be two possible reasons contributing to the decline in selectivity: a shortened transport route of the penetrating components and more undetected defects in the membranes. The undetected effects (even by SEM) would occur since GO nanosheets cannot be neglected compared to the bulk of the membrane. These factors might also be responsible for the observed decline in selectivity at high loading of GO.
Effect of H2O2 treatment
In order to further increase compatibility between graphene and the CS–PVP chains, GO nanosheets were treated with H2O2, which was found to enhance the oxygen content of GO from C10O0.78H0.38 to C10O0.99H0.73 (as per elemental analysis). As depicted in Fig. 9, the presence of additional oxygen functionalities seems to be instrumental in obtaining 40% higher membrane selectivity along with a slight improvement in total permeability of the membranes. One of the main reasons for the increased membrane performance after incorporation of GO is the presence of ‘enhanced oxygen functionality’ on the graphene surface, which would enhance both sorption and diffusion selectivity of the membrane. | ||
| Fig. 9 Comparison of membrane performance (selectivity and permeability) of CS–PVP-2.5 and CS–PVP-2.5 (U) for the water–EtOH mixture. | ||
Upon increasing the number of oxygen functionalities, the interaction of graphene with water molecules will also increase along with an improvement in interface interactions with the polymer chains as per the interaction model proposed in Fig. 10. Therefore, oxygen functionality of GO is responsible to improve the interface interaction with polymer chains; this can be evidenced by the improved values for sorption and diffusion selectivity of water.
 | ||
| Fig. 10 The interaction models showing the effect of H2O2 treatment. | ||
Effect of operating temperature
Fig. 11a displays the effect of the operating temperature on PV performance typically in the case of the CS–PVP-2 membrane for the 10 wt% water containing feed mixture. The operating temperature ranges of this study correspond to reasonable operating conditions of the PV process for ethanol dehydration. The permeability values increased from 99 to 114 Barrer, but the selectivity declined from 624 to 405. The increase in permeability at the loss of selectivity with temperature could be due to three reasons. One is that the increased mobility of the penetrating molecules in the bulk feed solution might have resulted in higher partial vapour pressure providing a higher driving force for permeating molecules, and this would facilitate transport of both liquid components in the membrane. The second is that flexibility of the blend polymer segments becomes enhanced at rising temperature thus accelerating non selective diffusion of the components. In the case of the GO-loaded membranes, variation of the interface between the polymer blend and GO induced by rising temperature would also play a major role. | ||
| Fig. 11 Variation of permeability and selectivity of the CS–PVP-2 membranes with (a) temperature at 10 wt% of water in EtOH and (b) feed composition at 30 °C. | ||
In other words, enhanced thermal mobility of the polymer segments at higher temperature would eventually create larger free volume in the matrix. As the free volume increases, permeation of water molecules along with EtOH also increases, thus exhibiting non-selective mass transport. The temperature dependent permeation and diffusion values exhibit Arrhenius trends that are quantified through calculation of the activation energy parameters for the temperature-dependent values of diffusion and permeability.
Effect of feed concentration
Fig. 11b demonstrates the effect of the feed concentration on PV dehydration performance of the CS–PVP-2 membranes, wherein it is observed that an increase in feed water composition increased the permeability at the cost of selectivity. This could be mainly attributed to an enhancement in the driving force followed by an increase in membrane swelling. An increase in feed water composition would result in an increase of water molecules on the feed side, while on the permeate side it is being constantly removed, leading to an increase of the chemical potential gradient (driving force). The increase in equilibrium swelling with increasing feed water composition is responsible for the increased membrane swelling, allowing for the easier permeation of both water and EtOH molecules.Arrhenius activation energy calculations
Arrhenius activation energy for permeability and diffusion processes of the feed mixture components have been calculated as:
 | (7) |
Dw vs. 1/T (not displayed to avoid overcrowding of figures) and from the least-squares fitting of the linear plots, activation energy values (kJ mol−1) for the permeability of water (Epw) and EtOH (EpEtOH) were calculated. Similarly, activation energy values for the diffusion of water (EDw) and EtOH (EDEtOH) were calculated. These data compiled in Table 2 show lower diffusion energy barrier values for water compared to EtOH. After incorporating GO into the CS–PVP matrix, the energy barrier for water declined, while that of EtOH increased, suggesting a water selective nature of the membranes. The decrease in the water diffusion energy barrier can be observed up to 2.5 wt% GO loading, and above this limit, the trend is reversed, due to possible particle agglomeration. On the other hand, the H2O2 treated GO loaded membranes showed much lower energy barrier values for water permeation than the untreated graphene loaded membranes.
| Membranes | Epw | EDw | ΔHs | EpEtOH | EDEtOH | ΔHs |
|---|---|---|---|---|---|---|
| Nascent CS–PVP | 14.6 | 24.5 | −9.9 | 35.2 | 47.0 | −11.8 |
| CS–PVP-1 | 11.8 | 23.2 | −11.4 | 39.3 | 51.4 | −12.1 |
| CS–PVP-2 | 10.2 | 22.0 | −11.8 | 40.8 | 54.3 | −13.5 |
| CS–PVP-2.5 | 8.1 | 19.8 | −11.7 | 44.6 | 57.9 | −13.3 |
| CS–PVP-3 | 9.4 | 21.4 | −12.0 | 42.0 | 55.2 | −13.2 |
| CS–PVP-2.5 (U) | 8.8 | 20.6 | −11.8 | 43.2 | 56.4 | −13.2 |
Heat of sorption values were calculated from the difference values of energy of activation for permeability and diffusion using:
| ΔHs = Epi − EDi | (8) |
The calculated ΔHs values presented in Table 2 suggest the nature of the transport phenomenon, which involves the combined effect of Henry’s and Langmuir’s type of sorption phenomenon.47 Henry’s law implies that the heat of sorption follows an endothermic process for liquid transport, leading to dissolution of the chemical species into that site within the membrane matrix. On the other hand, Langmuir’s sorption requires pre-existence of a site into which sorption occurs by a hole-filling mechanism to offer an exothermic contribution. The ΔHs values obtained are negative (exothermic contribution) for all membranes, indicating that Langmuir’s type of sorption is predominant in the present systems.
Membrane–solvent interactions
The selective mass transport through a dense polymeric membrane occurs through the combined effect of differential diffusion as well as physico-chemical interactions between liquid permeate molecules and the polymer matrix due to sorption. The sorption phenomenon is a thermodynamic process, which determines how much liquid is accommodated per unit volume into a polymer matrix. Equilibrium swelling data are used to calculate the Flory–Huggins interaction parameter (χip) between the polymer and the liquid molecules using the following equation:48|
lnαi = lnϕi + (1 − ϕi) + χip(1 − ϕi)2
| (9) |
 | (10) |
The membrane density (ρm) was calculated using the benzene-displacement method18:
 | (11) |
 | (12) |
The parameter χip represents the chemical compatibility between liquid molecules, i (water or EtOH) and the membrane polymer, p.
By incorporating the χip values, the membrane solubility parameter (δ) was calculated as:
 | (13) |
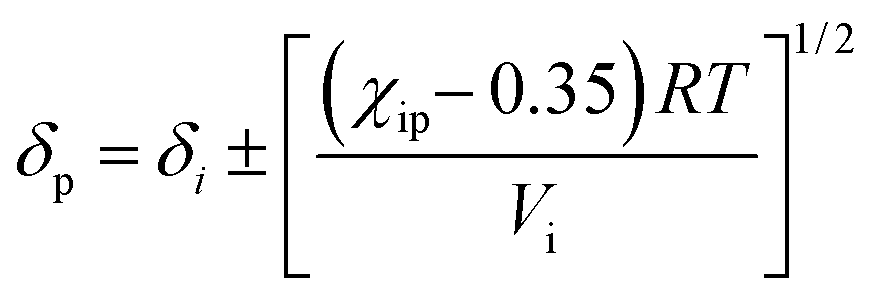 | (14) |
Since sorption of the membrane was measured in EtOH and water separately, we have chosen the one that matched better for the determination (here, i refers to water). Eqn (14) has two roots and hence, δp theoretically takes either of the two values while determining membrane–solvent solubility.
The lower χip values represent a stronger interaction between liquid media and the polymer. The results of Table 3 show almost ten-times lower χ1p (water–polymer interaction) values than χ2p (EtOH–polymer interaction), confirming the water-selective nature of the membranes. Notice that by incorporating GO nanoparticles, the χ1p values have decreased dramatically, while the χ2p values increased slightly. Such an effect is attributed to the added water-selective nature of the GO nanosheets, which was also confirmed by the water contact angle and equilibrium swelling data. By increasing the GO loading, the χ1p values decrease up to 2.5 wt% of loading, above which the trend is reverted, due to attainment of the saturation limit of GO in the membrane matrix. Comparatively, H2O2 treated GO loaded membranes have shown lower χ1p values than the corresponding untreated GO loaded membranes, due to an increase in hydrophilicity of GO after its H2O2 treatment. Similarly, the δp values show an increase with increased loading of the GO nanosheets, indicating increased hydrophilicity of the membrane. The δp values of the H2O2 treated GO loaded membranes showed higher values of δp than those of the unmodified GO-loaded membranes, which is due to the overall enhancement of the polar nature (hydrophilicity) of the membranes.
| Membranes | F–H parameters | δp (J1/2 cm−3/2) | Mc (g mol−1) | Ve × 103 (mol cm−3) | |
|---|---|---|---|---|---|
| χ1p | χ2p | ||||
| Nascent CS–PVP | −19 | −3.7 | 100 | 474 | 3.2 |
| CS–PVP-1 | −29 | −3.4 | 112 | 262 | 6.2 |
| CS–PVP-2 | −34 | −3.2 | 117 | 222 | 7.6 |
| CS–PVP-2.5 | −37 | −2.9 | 121 | 199 | 8.6 |
| CS–PVP-3 | −35 | −3.2 | 119 | 227 | 7.8 |
| CS–PVP-2.5 (U) | −34 | −3.0 | 118 | 221 | 7.5 |
Molar mass between cross-links and cross-link density
The molar mass between cross-link (Mc) and cross-link density (ve) of the polymers were calculated as:50
 | (15) |
 | (16) |
Usually, cross-linked polymers show higher ve and lower Mc values. As per the data given in Table 3, decreasing Mc values and increasing ve occur with increased GO loading, indicating enhanced membrane packing density, which could be due to the presence of the polar groups on both the CS/PVP blend and GO, leading to multiple H-bonded sites (interaction model). The increase in packing density was observed up to 2.5 wt% of GO loading and above this limit the trend reverted, since membranes reach a saturation limit at 2.5 wt% loading. On the other hand, H2O2 treated GO loaded CS/PVP blend nanocomposite membranes show higher cross-link densities than the unmodified GO loaded membranes as a result of improved interfacial compatibility between the GO nanosheets and the CS/PVP blend polymer after H2O2 treatment of GO.
Diffusion coefficient
Diffusion is a kinetic parameter, which determines the rate at which liquid molecules diffuse through the membrane. Diffusion coefficient (concentration independent) can be calculated as:
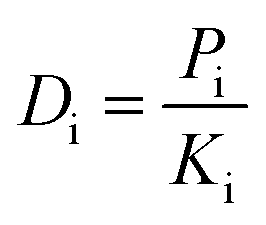 | (17) |
 | (18) |
Here, Ci stands for the liquid concentration inside the membrane and pi is the partial pressure of the i component.
All the membranes show (see Table 1) two orders of magnitude higher diffusion selectivity towards water molecules than towards EtOH molecules. With an increase in GO loading, diffusion of water molecules increased, leading to a decrease in the permeation of EtOH molecules. The tortuous diffusion pathways created in the matrix could facilitate permeation of the smaller water molecules compared to EtOH. The increasing trend of water diffusion is below 2.5 wt% GO loading and after this limit, the trend was reverted. Comparatively, the nanocomposite membranes containing H2O2 treated GO show better diffusion selectivity than that of the untreated graphene sheets containing membranes. The feed parameters, such as increase of water composition and temperature, have an adverse effect on the diffusion selectivity of the membranes.
Conclusions
One of the main concerns of graphene composite membranes for PV application is their low compatibility with a polymer matrix. In this work, we have attempted to address this issue by using H2O2 treated graphene incorporated into a CS/PVP blend matrix and these membranes, when tested for PV dehydration of ethanol as a function of GO loading, feed composition and feed temperature, showed improved results. The H2O2 treatment of GO enhanced the number of oxygen functionalized polar sites on the graphene surface, which has enhanced sorption and diffusion selectivity. The FE-SEM micrographs combined with XRD confirmed the good interface compatibility between the GO nanosheets and CS/PVP blend polymer. FTIR spectra proved enhanced H-bonding interactions, while contact angle studies proved an increase in hydrophilicity of the membranes with increased GO loading. TGA and DSC studies proved an enhancement of membrane thermal stability with increasing GO loading. The interaction parameter (χ) and diffusion coefficient values supported the progressive improvement of selectivity and permeability data with GO loading. Membranes attained an optimum concentration at 2.5 wt% loading, but any further increase of GO loading resulted in a decrease of the membrane performance. Overall, the H2O2 treatment proved to be effective to enhance PV performance of the nanocomposite membranes.Acknowledgements
Financial assistance from Department of Atomic Energy (DAE), Board of Research in Nuclear Sciences (BRNS) (Grant 2013/34/4-BRNS), Admar Mutt Education Foundation (AMEF), Bangalore, Dr K. Venkatesh, Director, CET, Jain University are gratefully acknowledged. Dr Suhas D. P. is thankful to Council for Scientific and Industrial Research for awarding a Senior Research Fellowship (F.N09/1052(0004)2K14-EMR-I).References
- D. Tilman, J. Fargione, B. Wolff, C. D’Antonio, A. Dobson, R. Howarth, D. Schindler, W. H. Schlesinger and D. Simberloff, Science, 2001, 292, 281–284 CrossRef CAS.
- D. Tilman, K. G. Cassman, P. A. Matson, R. Naylor and S. Polasky, Nature, 2002, 418, 671–677 CrossRef CAS PubMed.
- REN21, Renewables 2012 Global Status Report, REN21 Secretariat, 2012.
- C. N. Hamelinck, G. Van Hooijdonk and A. P. C. Faaij, Biomass Bioenergy, 2005, 28, 84–410 CrossRef.
- L. R. Lynd, Rev. Energy Environ., 1996, 21, 403–465 CrossRef.
- S. D. Bhat and T. M. Aminabhavi, Microporous Mesoporous Mater., 2006, 91, 206–214 CrossRef CAS.
- S. D. Bhat, B. V. K. Naidu, G. V. Shanbag, S. B. Halligudi and T. M. Aminabhavi, Sep. Purif. Technol., 2006, 49, 56–63 CrossRef CAS.
- B. Bolto, M. Hoang and Z. Xie, Chem. Eng. Process., 2011, 50, 227–235 CrossRef CAS.
- D. Anjali Devi, B. Smitha, S. Sridhar and T. M. Aminabhavi, J. Membr. Sci., 2006, 280, 45–53 CrossRef.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- S. S. Jawalkar, S. G. Adoor, M. Sairam, N. N. Mallikarjuna and T. M. Aminabhavi, J. Phys. Chem. B, 2005, 109, 15611–15620 CrossRef CAS PubMed.
- T. Uragami, K. Okazaki, H. Matsugi and T. Miyata, Macromolecules, 2002, 35, 9156–9163 CrossRef CAS.
- V. T. Magalad, G. S. Gokavi, C. Ranganathaiah, M. H. Burshe, C. Han, D. D. Dionysiou, M. N. Nadagouda and T. M. Aminabhavi, J. Membr. Sci., 2013, 430, 321–329 CrossRef CAS.
- S. D. Bhat and T. M. Aminabhavi, Sep. Purif. Technol., 2006, 51, 85–94 CrossRef CAS.
- S. G. Adoor, B. Prathab, L. S. Manjeshwar and T. M. Aminabhavi, Polymer, 2007, 48, 5417–5430 CrossRef CAS.
- S. G. Adoor, M. Sairam, L. S. Manjeshwar, K. V. S. N. Raju and T. M. Aminabhavi, J. Membr. Sci., 2006, 285, 182–195 CrossRef CAS.
- D. P. Suhas, T. M. Aminabhavi and A. V. Raghu, Appl. Clay Sci., 2014, 101, 419–429 CrossRef CAS.
- V. T. Magalad, G. S. Gokavi, K. V. S. N. Raju and T. M. Aminabhavi, J. Membr. Sci., 2010, 354, 150–161 CrossRef CAS.
- D. P. Suhas, T. M. Aminabhavi and A. V. Raghu, Polym. Eng. Sci., 2014, 54, 1774–1782 CAS.
- D. Y. Kang, H. M. Tong, J. Zang, R. P. Choudhury, D. S. Sholl, H. W. Beckham, C. W. Jones and S. Nair, ACS Appl. Mater. Interfaces, 2012, 4, 965–976 CAS.
- C. H. Kang, Y. F. Lin, Y. S. Huang, K. L. Tung, K. S. Chung, J. T. Chen, W. S. Huang, K. R. Lee and J. Y. Lai, J. Membr. Sci., 2013, 438, 105–111 CrossRef CAS.
- G. Liu, D. Yang, Y. Zhu, J. Ma, M. Nie and Z. Jiang, Chem. Eng. Sci., 2011, 66, 4221–4228 CrossRef CAS.
- Y. Shirazi and T. Mohammadi, Sep. Sci. Technol., 2013, 48, 716–727 CrossRef CAS.
- D. P. Suhas, A. V. Raghu, H. M. Jeong and T. M. Aminabhavi, RSC Adv., 2013, 3, 17120–17130 RSC.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- Z. Jia and Y. Wang, J. Mater. Chem. A, 2015, 3, 4405–4412 CAS.
- X. Chang, Z. Wang, S. Quan, Y. Xu, Z. Jiang and L. Shao, Appl. Surf. Sci., 2014, 316, 547–548 CrossRef.
- L. Shao, X. Cheng, Z. Wang, J. Ma and Z. Guo, J. Membr. Sci., 2014, 452, 82–89 CrossRef CAS.
- H. M. Hegab and L. Zou, J. Membr. Sci., 2015, 484, 95–106 CrossRef CAS.
- L. Shao, X. Chang, Y. Zhang, Y. Huang, Y. Yao and Z. Guo, Appl. Surf. Sci., 2013, 280, 989–992 CrossRef CAS.
- C.-H. Tsou, Q.-F. An, S.-C. Lo, M. D. Guzman, W.-S. Hung, C.-C. Hu, K.-R. Lee and J.-Y. Lai, J. Membr. Sci., 2015, 477, 93–100 CrossRef CAS.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- K. Huang, G. Liu, Y. Lou, Z. Dong, J. Shen and W. Jin, Angew. Chem., 2014, 53, 6929–6932 CrossRef CAS PubMed.
- D. P. Suhas, A. V. Raghu, H. M. Jeong and T. M. Aminabhavi, Ind. Eng. Chem. Res., 2014, 53, 14474–14484 CrossRef.
- J. T. Choi, T. D. Dao, K. M. Oh, H.-il. Lee, H. M. Jeong and B. K. Kim, Smart Mater. Struct., 2012, 21, 75017 CrossRef.
- T. M. Aminabhavi and R. S. Khinnavar, Polymer, 1993, 34, 1006–1018 CrossRef CAS.
- T. M. Aminabhavi and H. G. Naik, J. Appl. Polym. Sci., 2002, 83, 244–258 CrossRef CAS.
- R. W. Baker, J. G. Wijmans and Y. Huang, J. Membr. Sci., 2010, 348, 346–352 CrossRef CAS.
- V. T. Magalad, G. S. Gokavi, N. N. Mallikarjuna, B. Prathab and T. M. Aminabhavi, J. Phys. Chem. C, 2011, 115, 14731–14744 CAS.
- S. G. Adoor, S. D. Bhat, D. D. Dionysiou, M. N. Nadagouda and T. M. Aminabhavi, RSC Adv., 2014, 4, 52571–52582 RSC.
- J. G. Wijmans, J. Membr. Sci., 2003, 220, 1–3 CrossRef CAS.
- M. J. Holmes and M. V. Winkle, Ind. Eng. Chem., 1970, 62, 21–31 CrossRef CAS.
- G. Wang, B. Wang, J. Park, J. Yang, X. Shen and J. Yao, Carbon, 2009, 47, 68–72 CrossRef CAS.
- T. M. Aminabhavi, R. S. Khinnavar, S. B. Hargoppad, U. S. Aithal, Q. T. Nguyen and K. C. Hasan, J. Macromol. Sci., Chem., 1994, 34, 139–204 CrossRef.
- D. H. Weinkauf and D. R. Paul, ACS Symp. Ser., 1990, 423, 61–91 CrossRef.
- T. M. Aminabhavi and P. Munk, Macromolecules, 1979, 12, 607–613 CrossRef CAS.
- B. Prathab, V. Subramanian and T. M. Aminabhavi, Polymer, 2007, 48, 409–416 CrossRef CAS.
- G. M. Bristow and W. F. Watson, Trans. Faraday Soc., 1958, 54, 1731–1741 RSC.
- N. L. Le and T.-S. Chung, J. Membr. Sci., 2014, 454, 62–73 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |