
Monocyclic and bicyclic CO4: how stable are they?†
Abstract
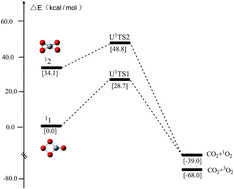
Seeking promising molecular species with huge energy release and significant kinetic stability continues to be a hot topic and a great challenge in the field of high-energy density materials (HEDMs). CO4 is the first high-order carboxide that has the potential as an energetic molecule. However, the intrinsic kinetic stability of its two most studied energy-rich isomers, i.e., 11 (monocyclic) and 12 (bicyclic), has remained quite unclear in spite of numerous studies. This has greatly hindered the quantitative stability assessment of 11 and 12 under various conditions as well as the justification of their prospect as energetic candidates. In this work, for the first time we report the rate-determining transition states associated with the CO2-elimination from 11 and 12. The thermodynamics of 11 and 12 was described using G3B3, CBS-QB3, G4, W1BD, CCSD(T)/CBS and CASPT2/CBS, while the kinetic stability was analyzed based on broken-symmetry UCCSD(T)/CBS and CASPT2/CBS single-point energy calculations on UB3LYP geometries. The rate-determining barriers for the dissociation of 11 and 12 into CO2 + 1O2 at 298 K were found to amount to 28.7 and 14.7 kcal mol−1 at the CASPT2(18e,12o)/CBS level of theory, and 23.5 and 21.1 kcal mol−1 at the UCCSD(T)/CBS level of theory, respectively. 11 is a kinetically stable energetic molecule, which releases 45.2 kcal mol−1 upon dissociation into CO2 + 1O2 at the CASPT2(18e,12o)/CBS level and 38.9 kcal mol−1 at the UCCSD(T)/CBS level, and could serve as a rigid energetic building block for larger oxocarbons. The bicyclic 12 releases much higher energy, 79.3 kcal mol−1 at the CASPT2(18e,12o)/CBS level and 73.4 kcal mol−1 at the CASPT2-corrected UCCSD(T)/CBS level whereas the barrier for dissociation is lower than that of monocyclic 11.
 Please wait while we load your content...
Please wait while we load your content...

