
Porphyrin chemodosimeters: synthesis, electrochemical redox properties and selective ‘naked-eye’ detection of cyanide ions†
Mandeep K. Chahal and
Muniappan Sankar*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: sankafcy@iitr.ac.in
First published on 9th November 2015
Abstract
Dicyanovinyl appended β-pyrrole substituted porphyrins have been synthesized and utilized as chemodosimeters for ratiometric and colorimetric sensing of cyanide ions. In addition to meso-tetraphenylporphyrin (TPP) based system, we have designed and synthesized octaphenylporphyrin (OPP) based moderately nonplanar systems. These systems also depicted effective π-conjugation between the pyrrole ring and its olefinic substituent which is reflected in the UV-vis absorption spectra as the red-shifting of both Soret and Q-bands and possibly the splitting of the absorption Soret band. This article describes the single crystal X-ray structure, electronic spectral and electrochemical redox properties of these porphyrin chemodosimeters. The stable anionic-porphyrin species formed after addition of CN− ions has a blue shifted unperturbed absorption spectrum due to the disruption of electronic resonance and charge-transfer interactions between the β-dicyanovinyl acceptor and porphyrin core. The probes exhibited excellent selectivity towards CN− over other anions such as F−, Cl−, Br−, NO3−, H2PO4−, AcO−, I−, HSO4−, ClO4− and PF6−. The action of the chemodosimeters were investigated by 1H-NMR titrations, ESI-mass spectrometry, electrochemical studies and DFT calculations which supported the formation of cyanide addition adduct (porphyrin–CN)−. These chemodosimeters depicted lowest detection limit (LOD) of 0.023–0.082 ppm, which is very much lower than the permissible limit (0.2 ppm) and other porphyrinoids reported till date. These porphyrins are also able to detect cyanide ions in (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) CH3CN:H2O mixture with low detection limits thereby providing practical applicability of these chemodosimeters.
:1, v/v) CH3CN:H2O mixture with low detection limits thereby providing practical applicability of these chemodosimeters.
Introduction
Porphyrins are biologically the most intriguing molecules after nucleic acids and proteins. They are essential in many vital processes that sustain life, for example, oxygen transport and storage, electron transport in redox processes, bio-catalysis and photosynthesis.1 Apart from biology, porphyrins and their metal complexes are being used in catalysis,2 dye-sensitized solar cells (DSSCs),3 photodynamic therapy (PDT),4 nonlinear optical (NLO)5 and self-organized materials6a and sorbents.6b In the last few years, the role of porphyrinoids in anion sensing has been explored extensively.7 Among the various anions known, cyanide is highly toxic; it binds strongly to iron in the heme and the active site of cytochrome c, which completely stops O2 transport in the blood and the mitochondrial electron-transport chain, thus inhibiting cellular respiration.8 So the continuous release of cyanide ions into the environment through its increased usage in various industrial applications like metallurgy, mining, electroplating, polymer synthesis, petrochemicals and organic synthesis, etc.9 is of great concern. Hence both biological and environmental aspects necessitate the development of cyanide-selective receptors. A wide variety of chemosensors10 to detect cyanide ions have been reported till date, however, they suffer from several limitations such as poor selectivity, especially in the presence of fluoride or acetate ions and absence of real life detection. For the purpose of minimizing the interference of other anions, many nucleophilic addition reaction mechanism based cyanide anion sensors have been reported. Among these cyanide sensors, vinyl-substituted derivatives display both selective and sensitive responses to various concentrations of cyanide anion.11 Chemosensors based on porphyrins, corrins and phthalocyanines for cyanide ions are well known.12 Of late, cyanide sensing through a chemodosimetric method by using a new calix[4]pyrrole derivative,13a Pd-calixphyrin13b and subphthalocyanine dye13c has been reported. Anzenbacher et al. prepared N-confused calix[4]pyrrole and regular calix[4]pyrrole based sensors for the detection of multianions using pyrrole macrocycle Knoevenagel chemistry.14A wide range of β-substituted porphyrins are known, but they all exhibit absorption spectra with common features i.e. red shifted Soret and Q-bands.15 Dicyanovinyl bearing β-substituted porphyrins have an effective π-conjugation and charge transfer between porphyrin macrocyclic ring and the peripheral substituent, resulting in perturbed absorption spectra. In this report, we have utilised this feature of red shifted, perturbed absorption spectra for selective cyanide ion sensing. In addition to the reported tetraphenylporphyrin (TPP) based system, we have designed and synthesized octaphenylporphyrin (OPP) based moderately nonplanar systems, which also depicted distorted electronic absorption spectra. Herein, we put forward two important observations of the porphyrin derivatives; in the first part, we highlight the efficient electronic coupling (resonance interaction) and charge-transfer between the acceptor and the porphyrin core in TPP and moderately nonplanar OPP-systems and in the second part, we describe how these porphyrins might be used as chemodosimetric sensors. We believe that this is the first successful attempt on β-substituted porphyrins being used as chemodosimeters (1–4) for selective, ‘naked-eye’ detection of cyanide ions in water-organic and organic media (Scheme 1).
 | ||
| Scheme 1 Synthetic scheme for the preparation of chemodosimeters 1–4. | ||
Results and discussion
Synthesis and characterization
Target porphyrins 1–4 were prepared by Knoevenagel condensation of β-formyl porphyrins and malononitrile in the presence of basic Al2O3 using modified literature methods (Fig. 1).16 2-Formyl-5,10,15,20-tetraarylporphyrin and 2-formyl-5,7,8,10,15,17,18,20-octaphenylporphyrin were prepared by Vilsmeier–Haack formylation of the respected copper(II) porphyrins using modified procedures as shown in Scheme S1 in the ESI.†17 These porphyrins were characterized by various spectroscopic techniques and single crystal XRD analyses (Fig. S1–S10 in the ESI†).To gain insight into effective π-conjugation and charge transfer between appended dicyanovinyl substituent and porphyrin core and also to check the effect of geometry of the porphyrin system on ion sensing, we have designed and synthesized olefinic acceptor appended moderately nonplanar octaphenylporphyrins (3 and 4). The representative absorption spectra of MTPP-X (M = Ni, 2H and X = CHO, MN) and MOPP-X (M = Ni, 2H and X = CHO, MN) in CH2Cl2 at 298 K are shown in Fig. S11 and S12 in the ESI and the corresponding spectral data is listed in Table S1.† All dicyanovinyl appended porphyrins exhibited characteristic split broad Soret bands (B bands). NiTPP-MN (1) exhibited both red shift (Δλmax = 24 nm) and blue shift (Δλmax = 42 nm) in the split Soret bands and red shift in Qx(0,0) band (Δλmax = 26 nm) relative to NiTPP-CHO. Similarly, NiOPP-MN (3) exhibited split Soret bands and red-shifted Qx(0,0) band. This red-shifting of both Soret and Q-bands and possibly the splitting of the absorption Soret band describe effective π-conjugation between the pyrrole ring and its olefinic substituent in OPP-system. Qx(0,0) band of dicyanovinyl appended porphyrins exhibited increments in molar extinction coefficients (ε) as compared to β-formyl substituted porphyrins. 1H NMR spectra of H2TTP-MN (2) and H2OPP-MN (4) exhibited proton resonances arising from the β-pyrrole, phenyl, dicyanovinyl and imino-NH protons. Up-fielded resonances of the β-pyrrole and meso-phenyl groups and down-fielded shift of the imino-NH protons (−1.57 ppm) for 4 in contrast to that of 2 (−2.52 ppm) in CDCl3 at 298 K were observed. The down-field shift of the imino protons of 4 is due to nonplanar conformation as observed in optimized geometries and UV-Vis spectrum in DMSO (Fig. S13 in the ESI†).18
The X-ray quality single crystals of 1 were obtained by direct diffusion of CH3OH into saturated CHCl3 solution of 1 and the crystallographic parameters are listed in Table S2 in the ESI.† ORTEP top and side views of 1 are shown in Fig. 1. Notably, 1 has shown ‘quasi planar conformation’ of the porphyrin macrocycle (ΔCβ = 0.38 Å and Δ24 = 0.21 Å) and the torsion angle between dicyanovinyl and pyrrole of porphyrin core is 26.39°. X-ray structure, optical absorption spectra and DFT studies (vide infra) strongly suggest that the dicyanovinyl acceptor on pyrrole β-carbon has nearly coplanar conformation with pyrrole ring of the porphyrin core. These observations demonstrate that the β-substituent exerts an appreciable electronic influence on the porphyrin π-electron system and provides a means of introducing charge-transfer character to prominent electronic transitions. Only abated solvatochromic shifts of the spectra were observed for these porphyrins due to partial localization of the pyrrole beta C–C double bond that partially blocks the π-conjugation over the whole porphyrin motif. Evidence for a charge-transfer process from the porphyrin ring is provided by the asymmetric shape and increased bandwidth of the B band (H2TPP-MN (FWHM = 55 nm) and H2OPP-MN (FWHM = 58 nm) with respect to H2TPP (FWHM = 14 nm)) of chromophores (1–4).19 Such an ICT contribution to the overall electronic structure has also been strongly suggested by DFT-calculations. The strong electronic resonance and charge transfer interaction between dicyanovinyl acceptor and the 18π-electron is responsible for spectral perturbation as compared to the parent β-formyl-substituted porphyrins. Our current porphyrin systems have the same olefinic acceptor-attached to pyrrole carbon, so varied spectral perturbation of the porphyrins observed herein can be attributed to the different peripheral crowding resulting in dissimilar extent of nonplanar deformation. The spectral perturbation is more for 1 and 2 as compared to 3 and 4, respectively, which is parallel to the order of quasi planar and nonplanar conformation of the porphyrin core.
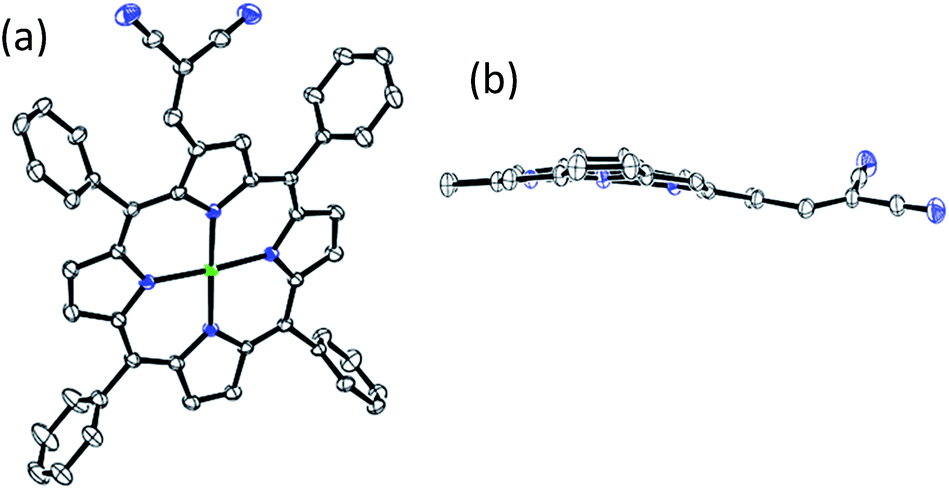 | ||
| Fig. 1 The ORTEP diagrams showing (a) top and (b) side views of NiTPP-MN (1). In side view, meso-phenyl groups are not shown for clarity. | ||
Electrochemical redox properties
To examine the effect of β-substituents and nonplanarity of the macrocycle on electrochemical redox potentials, we have carried out cyclic voltammetric studies. Cyclic voltammograms of 1–4 in CH2Cl2 containing 0.1 M TBAPF6 as supporting electrolyte are shown in Fig. 2. | ||
| Fig. 2 Cyclic voltammograms of 1–4 in CH2Cl2 containing 0.1 M TBAPF6 as the supporting electrolyte using Ag/AgCl as the reference electrode. | ||
Under similar conditions, the corresponding MTPP derivatives were also examined and the electrochemical redox data (vs. Ag/AgCl) is listed in Table 1. These porphyrins are oxidized in two one-electron steps and reduced in two one-electron steps. In case of nickel-metallated dicyanovinyl appended systems, a third oxidation step might be observed.20 1–4 exhibited 370–460 mV anodic shift in first ring reduction potentials in comparison to MTPP and MOPP (M = 2H, Ni(II)) due to the conjugative and electron-withdrawing nature of the β-dicyanovinyl substituents. Similarly, we found 120–200 mV anodic shift in the oxidation potentials of 1–4 as compared to MTPP/MOPP. As we increase the electron withdrawing strength of β-substituent from –CHO to –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (CN)2 group, the HOMO–LUMO gap decreases (0.18–0.32 V) as compared to MTPP/MOPP as shown in Table 1 and Fig. S15,† ESI due to extensive stabilization of LUMO. The electrochemical redox data of these porphyrins clearly indicate the electron deficient nature of porphyrin π-system.
(CN)2 group, the HOMO–LUMO gap decreases (0.18–0.32 V) as compared to MTPP/MOPP as shown in Table 1 and Fig. S15,† ESI due to extensive stabilization of LUMO. The electrochemical redox data of these porphyrins clearly indicate the electron deficient nature of porphyrin π-system.
| Porphyrin | Oxidation (V) | ΔE1/2 (V) | Reduction (V) | |||
|---|---|---|---|---|---|---|
| I | II | III | I | II | ||
| a Scan rate 0.1 V s−1. ΔE1/2 = I oxd. − I red. Pt working and Pt wire counter electrodes were used. | ||||||
| H2TPP | 1.00 | 1.34 | 2.23 | −1.23 | −1.54 | |
| H2(CHO)TPP | 1.05 | 1.26 | 2.04 | −0.99 | −1.25 | |
| H2OPP | 0.83 | 0.95 | 2.02 | −1.19 | ||
| NiTPP | 1.02 | 1.32 | 2.30 | −1.28 | −1.72 | |
| Ni(CHO)TPP | 1.10 | 1.31 | 2.20 | −1.10 | −1.44 | |
| NiOPP | 0.91 | 1.19 | 2.23 | −1.32 | −1.71 | |
| NiTPP-MN (1) | 1.15 | 1.32 | 1.89 | 2.03 | −0.88 | −1.22 |
| H2TPP-MN (2) | 1.15 | 1.39 | 1.92 | −0.77 | −1.04 | |
| NiOPP-MN (3) | 1.03 | 1.21 | 1.8 | 1.91 | −0.88 | −1.25 |
| H2OPP-MN (4) | 1.02 | 1.84 | −0.82 | −1.08 | ||
Cyanide ion sensing
The anion recognition properties of all synthesized porphyrins (1–4) were studied in toluene with various anions, using UV-Visible spectroscopy with the addition of the aliquot anion in the form of TBA salts. In all synthesized porphyrins, both Soret and Q-bands were blue shifted on addition of tetrabutylammonium cyanide and perturbation in optical spectra was lost as shown in Fig. 3a and S16–S18 in the ESI.† When 1 (8 μM) was titrated with CN− ions (0–1.87 × 10−5 M), intensity of the bands at 385 nm, 450 nm and 602 nm gradually decreased and new bands arose at 416 nm and 534 nm, with three isosbestic points at 392 nm, 431 nm and 549 nm, respectively. The colour of the solution changed rapidly from green to light pink (Fig. 3c). This hypsochromic-shift and loss of perturbation in optical spectra led to inhibition of both π-conjugation and charge-transfer between the dicyanovinyl and porphyrin core. Therefore, we expect the addition of one cyanide ion to the chemodosimeter via nucleophilic addition mechanism. Further, continuous variation method (Job's plot) invariably showed 1:1 binding stoichiometry (Fig. 3d and S19 in the ESI†).
 | ||
| Fig. 3 Absorption and visual changes of NiTPP-MN (1) due to the addition of anions in toluene. (a) UV/Vis titration of CN− (2.3 equiv.) to 1 (8 μM), (b) absorption spectra of 1 (8 μM) in the presence of different anions, (c) visual changes by addition of various anions to 1, (d) Job's plot of 1 with CN− ions. | ||
The electronic resonance interaction and the charge transfer between the β-substituent and the porphyrin core were disrupted by nucleophilic addition of CN− at α-carbon of the dicyanovinyl group and this implies its clear transformation into a new species. This serves as evidence in support of the claim that any substituent that causes perturbation of the porphyrin spectrum should have strong electronic resonance and charge transfer interactions with the porphyrin π-system in addition to de-symmetrization of the porphyrin geometry.
In order to evaluate the selective cyanide ion sensing of all the chemodosimeters, absorption spectral changes upon addition of excess of various anions including F−, Cl−, Br−, NO3−, H2PO4−, AcO−, I−, HSO4−, ClO4− and PF6− were studied. Characteristic spectral changes with rapid colour change were observed only for cyanide ions (Fig. 3b, c and S20–S22 in the ESI†). Interestingly, the most commonly interfering anions such as fluoride and acetate did not show any response at all. Competition experiments to ascertain the selectivity of the probes in the presence of the other interfering anions were performed in toluene. In these measurements, 10 equiv. of the other interfering anions (excess) were used (Fig. S23 in the ESI†). A representative selectivity bar chart observed for 1–4 is shown in Fig. 4. It clearly confirms the exclusive cyanide ion detecting ability of these porphyrins even in the presence of all interfering anions in the same solution. From the above titrations studies, it should be noted that relatively non planar OPP-system (3 and 4) also responds similarly for the detection of toxic cyanide ions. The reaction rate constant of 1 with CN− was estimated under a pseudo-first-order approximation as the cyanide anion was used in a high concentration. A plot of ln[(Amax − At)/Amax], where Amax and At are the absorbance at the end of the reaction and time, t respectively, against time gave a linear fit with the slope corresponding to the pseudo-first-order rate constant of 0.012 s−1 (Fig. S24 in the ESI†).
 | ||
| Fig. 4 Selectivity profile of porphyrin chemodosimeters with various anions. ΔA refers to A − A0, where A and A0 stand for the absorbance in the presence and absence of anions, respectively and X− represent mixture of all anions. | ||
In order to support the chemodosimetric approach for cyanide ion sensing, we monitored the 1H NMR spectral changes observed after the addition of cyanide ion in CDCl3 at room temperature. As shown in Fig. 5a in case of 1, the vinylic proton (Ha) shown at 7.25 ppm completely disappears upon addition of 1 equiv. of cyanide ions. A new signal appears at 4.2 ppm. The new signal is assignable to the tricyanoethyl proton. Also adjacent β-proton (Hb) is shifted up-field which also supports the formation of [1·CN]− adduct. All these observations are in accordance with the nucleophilic addition of cyanide at the π-carbon of the vinylic linkage. In all the porphyrin chemodosimeters, same NMR changes are observed as shown in Fig. S25–S27 in the ESI.† The 1:1 adduct is further confirmed by ESI-mass spectrometry analysis, where the adduct shows the molecular ion peak in the negative ion mode (Fig. 5b and S28 in the ESI†).
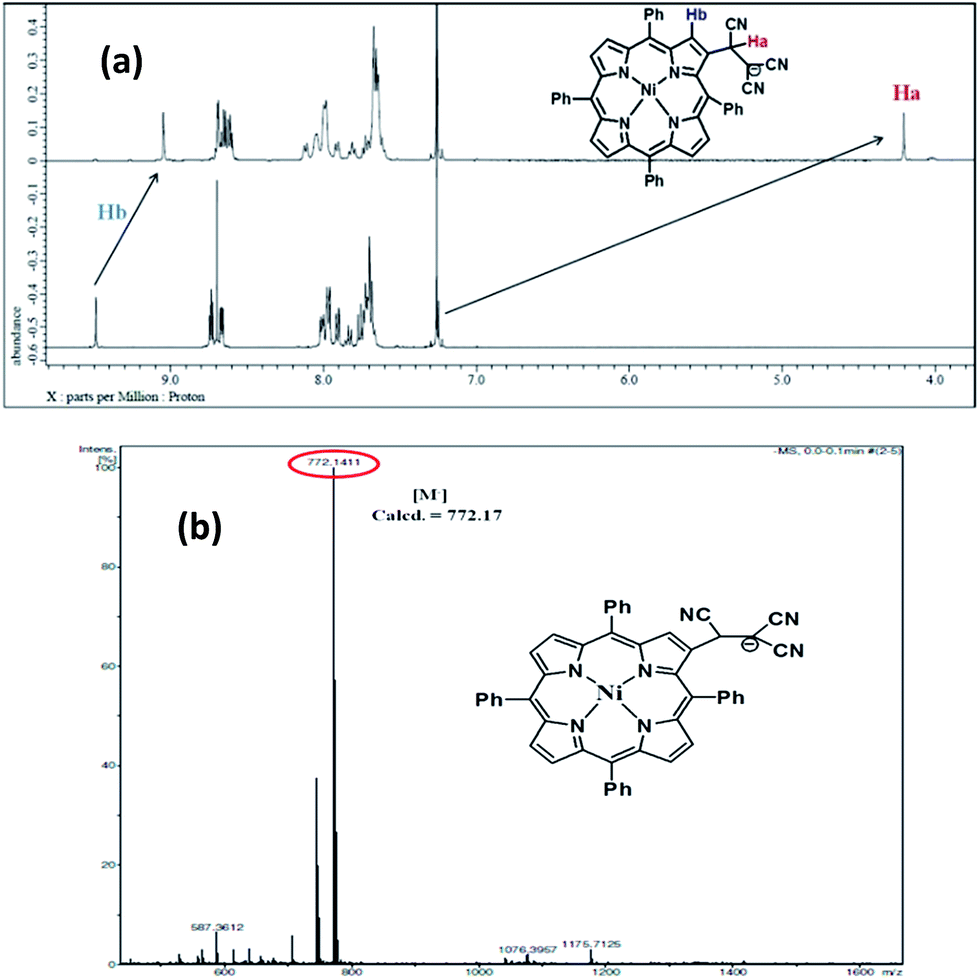 | ||
| Fig. 5 (a) 1H NMR spectra of the adduct [1·CN]− after addition of CN− ions (1 equiv.) to NiTPP-MN (1) (5 × 10−3 M) in CDCl3 at 298 K, (b) ESI-MS spectrum of adduct [1·CN]− after addition of CN− ions (1 equiv.) to NiTPP-MN (1). | ||
Nevertheless, in order to get further insight into the binding ability of these sensors 1–4 with CN− ions, we have carried out cyclic voltammetric (CV) and differential pulse voltammetric (DPV) studies of 1–4 with the addition of cyanide ions (Fig. 6, S29 and S30 in the ESI†). Interestingly, 1–4 exhibited 230–420 mV cathodic shift in first ring reduction potentials in the presence of CN− ions whereas oxidation remain unaltered clearly indicating the formation anionic species and interruption of π-conjugation. Table 2 represents the electrochemical first ring redox data of 1–4 in the presence and absence of CN− ions.
 | ||
| Fig. 6 (a) Cyclic voltammetric (b) DPV traces of 1 in absence and presence of CN− ions in CH2Cl2 containing 0.1 M TBAPF6 at 298 K. | ||
| Porphyrin | Porphyrin (without CN−) | Porphyrin (with CN−) | ||
|---|---|---|---|---|
| 1st ox | 1st red | 1st ox | 1st red | |
| 1 | 1.159 | −0.886 | 1.189 | −1.247 |
| 2 | 1.158 | −0.77 | 1.201 | −1.096 |
| 3 | 1.033 | −0.889 | 1.03 | −1.237 |
| 4 | 1.026 | −0.817 | 1.05 | −1.045 |
DFT studies
To gain insight into the electronic structure of the porphyrins and rationalize the changes in the optical properties induced by the reaction with cyanide, density functional theoretical computations were performed on the porphyrins and their cyanide adducts at the B3LYP/LANL2DZ level using the Gaussian 09 program suite.21 The β-pyrrole ring of porphyrin in 1 and 2 were nearly coplanar with dicyanovinyl unit (as shown in crystal structure). But the pyrrole ring in 3 was slightly tilted (38°) from the dicyanovinyl unit when compared to the pyrrole ring (23.5°) in 1 (Table S3 and Fig. S31, S32 in the ESI†). Overall the porphyrins were reasonably conjugated with the β-dicyanovinyl acceptors in all the systems. After the addition of CN− ions, the anionic porphyrin adducts were formed. In all anionic species the side arm stands nearly perpendicular to the pyrrole ring (torsion angle varied from 77.5° to 91.7°) thereby disrupting the interaction in all the systems (Table S3 and Fig. S33 in the ESI†). Also, in optimized structures of these chemodosimeters, HOMO resides on porphyrin while LUMO resides on dicyanovinyl arm establishing the charge transfer interaction in these donor–acceptor systems (Fig. S34 and S36 in the ESI†). The HOMO and LUMO switches after the addition of cyanide ion and is localized on the electron rich tricyanoethylate group supporting the formation of anionic porphyrin species (Fig. S35 and S37 in the ESI†). The change of HOMO and LUMO after the reaction with CN− is important for the elucidation of redox processes. Since in the (porphyrin·CN−) adduct the HOMO resides on electron rich tricyanoethylate group resulting in the cathodic shift in first ring reduction potentials supported by the cyclic voltammetric (CV) and differential pulse voltammetric (DPV) studies.Cyanide sensing in aqueous medium
To describe the practical applicability of 1–4, we tried to establish a suitable aqueous system. The porphyrin chemodosimeter detects cyanide ion in 10% H2O–CH3CN medium with appreciably low detection limits (Table S4 and Fig. S38–S40 in the ESI†). To get a quantitative idea of the sensing, the experiment was further performed by using CN− ions separately. Also, 1 and 3 exhibited ‘naked-eye’ colour change in presence of CN− ions in neutral water, though the time taken (1–2 min) to sense CN− ions was more as compared to organic media (1–2 seconds) (Fig. 7). The limits of detection (LOD) for CN− ions were calculated in toluene and H2O–CH3CN mixture. These chemodosimeters depicted lowest detection limit of 0.023 ppm in toluene, which is very much lower than the permissible limit (0.2 ppm)22 and other porphyrinoids till date.13 These porphyrins are able to detect CN− ions in solutions having concentrations as low as 1.64 ppm in H2O:CH3CN mixture. The generated anionic porphyrin species (Scheme 2) are very stable as the optical spectra remain unchanged after weeks and also 1H NMR and mass spectra remain unaffected. The hypsochromic shift in UV-Vis spectra rules out the possibility of axial ligation of cyanide to nickel in case of metallated systems12h and deprotonation of inner –NH protons in case of free base systems.10h
 | ||
| Fig. 7 Visual-colorimetric detection of CN− present in H2O (porphyrin dissolved in 10% H2O–MeCN and CN− present in neutral H2O). | ||
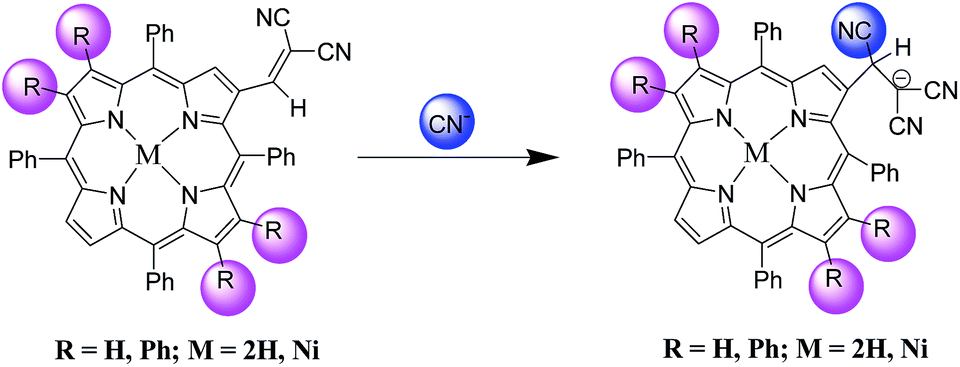 | ||
| Scheme 2 Proposed mechanism for the formation of stable anionic species with cyanide ions. | ||
Sensor strips and solid state detection
For the practical application of 1–4, test kits were prepared by immersing Whatman filter papers in toluene solution of 1–4 (1 mM) and then air dried. These test kits coated with 1 were exposed to different anion solutions (1 mM) in toluene for 1–2 seconds, the colour change from green to light pink was observed only with the CN− ion in toluene (Fig. 8a) whereas no colour change observed for other anions. Visual detection of trace amounts of cyanide ions is very convenient for prompt identification. For this purpose, dye 1 was added to CHCl3 solutions of different anions viz. CN−, F−, Cl−, Br−, NO3−, H2PO4−, AcO−, I−, HSO4−, ClO4− and PF6− (in different sample vials) and adsorbed on TLC plate at different spots. After running TLC in CHCl3, all spots have same Rf value except that containing CN− ions (Fig. 8b). Hence, test strips and TLC plates demonstrated the instant visualization and detection of trace amounts of cyanide ions. | ||
| Fig. 8 Photographs of test kits with 1 (1 mM) for detecting the cyanide ion in (a) toluene solution and (b) instant solid state detection of CN− ions. | ||
Conclusion
In summary, we have demonstrated efficient electronic and charge transfer communication between dicyanovinyl side arm and porphyrin macrocyclic core in both quasi planar and moderately nonplanar systems. We have successfully utilized these systems for ratiometric ‘naked-eye’ cyanide ion sensing and justified the chemodosimetric sensing property of the synthesized porphyrins. To the best of our knowledge, such type of cyano adduct formation is hitherto unknown in β-substituted porphyrin chemistry. The experimental results are in fair accordance with computational studies (DFT calculations) for elucidation of redox processes. In addition, these porphyrin-systems exhibit instant visualization of cyanide ions both in solid-state and using test strips.Experimental section
Materials
Pyrrole, benzaldehyde, Ni(OAc)2·4H2O, POCl3, DMF, 1,2-dichloroethane, basic Al2O3 and Na2SO4 were purchased from HiMedia, India and used as received. Malononitrile was purchased from Alfa Aesar and used as received. All solvents employed in the present work were of analytical grade and distilled or dried before use. Silica gel (100–200 mesh) was purchased from Rankem and used as received. TBAPF6 was recrystallised twice with ethanol and dried at 50 °C under vacuum for 3 days. The tetrabutylammonium salts (TBAX, X = CN−, F−, Cl−, Br−, I−, HSO4−, OAc−, H2PO4−, ClO4−, PF6− and NO3−) were purchased from Alfa Aesar and used as received. Dry CH2Cl2 for CV studies was distilled thrice from CaH2 and toluene (for UV-Visible spectral studies) was dried and distilled from a sodium–benzophenone mixture.Instrumentation and methods
Optical absorption spectra were recorded using an Agilent Cary 100 spectrophotometer using a pair of quartz cells 10 mm path length. 1H and 13C NMR spectra were recorded using a Bruker AVANCE 500 MHz and JEOL ECX 400 MHz spectrometers in CDCl3. MALDI-TOF-MS spectra were measured using a Bruker UltrafleXtreme-TN MALDI-TOF/TOF spectrometer using HABA as a matrix and ESI mass spectra were recorded on a Bruker Daltonics microTOF mass spectrometer using CH3CN as solvent. The ground state geometry optimisation was carried out by DFT calculations using B3LYP functional with LANL2DZ basis set using G09 program suite. Electrochemical measurements were carried out using CH instrument (CH 620E). A three electrode assembly was used consisted of a platinum working electrode, Ag/AgCl as a reference electrode and a Pt-wire as a counter electrode. All measurements were performed in triple distilled CH2Cl2 containing 0.1 M TBAPF6 as supporting electrolyte, which was degassed by argon gas purging. Cyanide detection studies were carried out in distilled toluene at 298 K. We calculated the limit of detection (LOD) from the titration experiments following the reported method.23 Single-crystal XRD data of 1 was collected on a Bruker Apex-II CCD diffractometer equipped with a liquid cryostat. The single crystals obtained were mounted on mounting loops. The diffraction data were collected using Bruker APEXII diffractometer at 293 K equipped with graphite-monochromated Mo Kα (λ = 0.71073 Å) by the ω–2θ scan.UV/Vis (CH2Cl2): λmax in nm (logε) 385(4.82), 451(5.18), 550(4.07), 606(4.36). 1H NMR in CDCl3 (400 MHz): δH (ppm) 9.489 (S, 1H, β-H), 8.747–8.657 (m, 6H, β-H), 8.02–7.9 (m, 8H, meso-o-phenyl-H), 7.857–7.666 (m, 12H, meso-m,p-phenyl-H), 7.247 (s, 1H); MALDI-TOF-MS (m/z): found 746.06 [M+], calcd 746.17. Anal. calcd for C48H28N6Ni: C, 77.13; H, 3.78; N, 11.24%. Found: C, 77.69; H, 3.69; N, 11.68%.
UV/Vis (CH2Cl2): λmax in nm (logε) 401(4.92), 453(5.24), 532(4.31), 580(3.97), 615(3.90), 675(3.98). 1H NMR in CDCl3 (500 MHz): δH (ppm) 9.526 (s, 1H, β-H), 8.99–8.736 (m, 6H, β-H), 8.249–8.132 (m, 8H, meso-o-phenyl-H), 7.944–7.766 (m, 12H, meso-m,p-phenyl-H), 7.282 (s, 1H), −2.522 (s, 2H, imino NH); ESI-MS (m/z): found 691.27 [M + H], calcd 691.26. Anal. calcd for C48H30N6: C, 83.46; H, 4.38; N, 12.17%. Found: C, 83.39; H, 4.80; N, 11.90%.
UV/Vis (CH2Cl2): λmax in nm (logε) 401(4.79), 465(5.20), 569(4.23), 627(4.32). 1H NMR in CDCl3 (500 MHZ): δH (ppm) 8.905 (s, 1H, β-H), 8.159–8.149 (m, 2H, β-H), 7.515–7.421 (m, 8H, meso-o-phenyl-H), 7.336–7.074 (m, 12H, meso-m,p-phenyl-H), 6.891–6.79 (m, 20H), 6.583 (s, 1H); MALDI-TOF-MS (m/z): found 1050.24 [M+], calcd 1050.29. Anal. calcd for C72H44N6Ni: C, 82.21; H, 4.22; N, 7.99%. Found: C, 82.56; H, 3.69; N, 7.68%.
UV/Vis (CH2Cl2): λmax in nm (logε) 469(5.20), 558(4.22), 608(3.99), 699(3.91). 1H NMR in CDCl3 (500 MHz): δH (ppm) 8.872 (s, 1H, β-H), 8.204 (s, 2H, β-H), 7.924–7.739 (m, 8H, meso-o-phenyl-H), 7.434–7.202 (m, 12H, meso-m,p-phenyl-H), 6.986–6.813 (m, 20H), 6.723 (s, 1H), −1.57 (s, 2H, imino NH); MALDI-TOF-MS (m/z): found 995.36 [M+], calcd 995.17. Anal. calcd for C72H46N6: C, 86.90; H, 4.66; N, 8.44%. Found: C, 86.67; H, 4.69; N, 8.98%.
Acknowledgements
We sincerely thank SERB, CSIR and BRNS, India for financial support. MKC thanks UGC, India for senior research fellowship.Notes and references
- (a) L. R. Milgrom, The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, New York, 1997 Search PubMed; (b) S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, 1994 Search PubMed.
- (a) S. P. de Visser, J. S. Valentine and W. Nam, Angew. Chem., Int. Ed., 2010, 49, 2099–2101 CrossRef CAS PubMed; (b) M. Araghi, V. Mirkhani, M. Moghadam, S. Tangestaninejad and I. Mohammdpoor-altork, Dalton Trans., 2012, 41, 3087–3094 RSC.
- (a) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed; (b) M. Urbani, M. Gratzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed; (c) T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–462 RSC.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- (a) M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. N. Mhuircheartaigh, Adv. Mater., 2007, 19, 2737–2774 CrossRef CAS; (b) A. Wang, L. Long, S. Meng, X. Li, W. Zhao, Y. Song, M. P. Cifuentes, M. G. Humphrey and C. Zhang, Org. Biomol. Chem., 2013, 11, 4250–4257 RSC; (c) M. Zawadzka, J. Wang, W. J. Blau and M. O. Senge, Photochem. Photobiol. Sci., 2013, 12, 996–1007 RSC.
- (a) C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS PubMed; (b) K. S. Suslick, P. Bhyrappa, J. H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283–291 CrossRef CAS PubMed.
- (a) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC Publishing, Cambridge, UK, 2006 Search PubMed; (b) P. Anzenbacher, R. Nishiyabu and M. A. Palacois, Coord. Chem. Rev., 2006, 250, 2929–2938 CrossRef CAS; (c) S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525–2536 CrossRef CAS PubMed; (d) P. A. Gale and C.-H. Lee, Top. Heterocycl. Chem., 2010, 24, 39–73 CAS; (e) F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 16138–16146 CrossRef PubMed; (f) L. C. Gilday, N. G. White and P. D. Beer, Dalton Trans., 2012, 41, 7092–7097 RSC; (g) J. M. M. Rodrigues, A. S. F. Farinha, P. V. Muteto, S. M. Woranovicz-Barreira, F. A. A. Paz, M. G. P. M. S. Neves, J. A. S. Cavaleiro, A. C. Tome, M. T. S. R. Gomes, J. L. Sessler and J. P. C. Tome, Chem. Commun., 2014, 50, 1359–1361 RSC; (h) Y. Kubo, M. Yamamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 42, 2036–2040 CrossRef CAS PubMed; (i) M. Takeuci, T. Shioya and T. M. Swager, Angew. Chem., Int. Ed., 2001, 40, 3372–3376 CrossRef.
- B. Vennesland, E. E. Comm, C. J. Knownles, J. Westly and F. Wissing, Cyanide in Biology, Academic Press, London, 1981 Search PubMed.
- C. Young, L. Tidwell and C. Anderson, Cyanide: Social, Industrial, and Economic Aspects, Minerals, Metals, and Materials Society, Warrendale, 2001 Search PubMed.
- (a) Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC; (b) P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 Search PubMed; (c) N. Kumari, S. Jha and S. Bhattacharya, J. Org. Chem., 2011, 76, 8215–8222 CrossRef CAS PubMed; (d) A. K. Mahapatra, S. K. Manna, B. Pramanik, K. Maiti, S. Mondal, S. S. Alia and D. Mandal, RSC Adv., 2015, 5, 10716–10722 RSC; (e) C. Wang, J. Jia, W.-N. Zhang, H.-Y. Zhang and C.-H. Zhao, Chem.–Eur. J., 2014, 20, 16590–16601 CrossRef CAS PubMed; (f) Q. Lin, T.-T. Lu, X. Zhu, B. Sun, Q.-P. Yang, T.-B. Weia and Y.-M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC; (g) Y. Sun and S. Wang, Inorg. Chem., 2010, 49, 4394–4404 CrossRef CAS PubMed; (h) N. Chaudhri and M. Sankar, RSC Adv., 2015, 5, 3269–3275 RSC.
- (a) Z. Liu, X. Wang, Z. Yang and W. He, J. Org. Chem., 2011, 76, 10286–10290 CrossRef CAS PubMed; (b) J.-L. Fillaut, H. Akdas-Kilig, E. Dean, C. Latouche and A. Boucekkine, Inorg. Chem., 2013, 52, 4890–4897 CrossRef CAS PubMed; (c) W. Chen, Z. Zhang, X. Li, H. Ǻgren and J. Su, RSC Adv., 2015, 5, 12191–12201 RSC; (d) R. K. Konidena and K. R. J. Thomas, RSC Adv., 2014, 4, 22902–22910 RSC; (e) G. R. Kumar and P. Thilagar, Dalton Trans., 2014, 43, 7200–7207 RSC.
- (a) K.-I. Hong, H. Yoon and W.-D. Jang, Chem. Commun., 2015, 51, 7486–7488 RSC; (b) Y.-H. Kim and J.-I. Hong, Chem. Commun., 2002, 512–513 RSC; (c) J. L. Worlinsky, S. Halepas and C. Brückner, Org. Biomol. Chem., 2014, 12, 3991–4001 RSC; (d) C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272–1274 CrossRef PubMed; (e) F. H. Zelder, Inorg. Chem., 2008, 47, 1264–1266 CrossRef CAS PubMed; (f) L. D. Chen, X. U. Zou and P. Bühlmann, Anal. Chem., 2012, 84, 9192–9198 CAS; (g) H. Liu, X. B. Shao, M. X. Jia, X. K. Jiang, Z. T. Li and G. J. Chen, Tetrahedron, 2005, 61, 8095–8100 CrossRef CAS; (h) R. Kumar, N. Chaudhri and M. Sankar, Dalton Trans., 2015, 44, 9149–9157 RSC.
- (a) S.-J. Hong, J. Yoo, S.-H. Kim, J. S. Kim, J. Yoon and C.-H. Lee, Chem. Commun., 2009, 189–191 RSC; (b) M. G. D. Holaday, G. Tarafdar, B. Adinarayana, M. L. P. Reddy and A. Srinivasan, Chem. Commun., 2014, 50, 10834–10836 RSC; (c) E. Palomares, M. V. Martinez-Diaz, T. Torres and E. Coronado, Adv. Funct. Mater., 2006, 16, 1166–1170 CrossRef CAS.
- (a) R. Nishiyabu, M. A. Palacios, W. Dehaen and P. Anzenbacher Jr, J. Am. Chem. Soc., 2006, 128, 11496–11504 CrossRef CAS PubMed; (b) R. Nishiyabu and P. Anzenbacher Jr, J. Am. Chem. Soc., 2005, 127, 8270–8271 CrossRef CAS PubMed; (c) R. Nishiyabu and P. Anzenbacher Jr, Org. Lett., 2006, 8, 359–362 CrossRef CAS PubMed; (d) M. A. Palacios, R. Nishiyabu, M. Marquez and P. Anzenbacher Jr, J. Am. Chem. Soc., 2007, 129, 7538–7544 CrossRef CAS PubMed; (e) Y. Liu, T. Minami, R. Nishiyabu, Z. Wang and P. Anzenbacher Jr, J. Am. Chem. Soc., 2013, 135, 7705–7712 CrossRef CAS PubMed.
- (a) M. J. Crossley, M. M. Harding and C. W. Tansey, J. Org. Chem., 1994, 59, 4433–4437 CrossRef CAS; (b) K. M. More, S. S. Eaton and G. R. Eaton, Inorg. Chem., 1981, 20, 2641–2647 CrossRef CAS; (c) J. E. Baldwin, M. J. Crossley and J. DeBernardis, Tetrahedron, 1982, 38, 685–692 CrossRef CAS; (d) P. Bhyrappa, M. Sankar and B. Varghese, Inorg. Chem., 2006, 45, 4136–4149 CrossRef CAS PubMed.
- C.-T. Chen, H.-C. Yeh, X. Zhang and J. Yu, Org. Lett., 1999, 1, 1767–1770 CrossRef CAS.
- E. E. Bonfantini, A. K. Burrell, W. M. Campbell, M. J. Crossley, J. J. Gosper, M. M. Harding, D. L. Officer and D. C. W. Reid, J. Porphyrins Phthalocyanines, 2002, 6, 708–719 CrossRef CAS.
- (a) C. J. Medforth and K. M. Smith, Tetrahedron Lett., 1990, 31, 5583–5586 CrossRef CAS; (b) M. O. Senge, Chem. Commun., 2006, 243–256 RSC.
- (a) E. Annoni, M. Pizzotti, R. Ugo, S. Quici, T. Morotti, M. Bruschi and P. Mussini, Eur. J. Inorg. Chem., 2005, 3857–3874 CrossRef CAS; (b) J. H. Fuhrhop, Struct. Bonding, 1974, 18, 1–67 CrossRef CAS; (c) J. C. Earles, K. C. Gordon, A. W. I. Stephenson, A. C. Partridge and D. L. Officer, Phys. Chem. Chem. Phys., 2011, 13, 1597–1605 RSC.
- (a) Y. Fang, M. O. Senge, E. V. Caemelbecke, K. M. Smith, C. J. Medforth, M. Zhang and K. M. Kadish, Inorg. Chem., 2014, 53, 10772 CrossRef CAS PubMed; (b) S. Richeter, C. Jeandon, J.-P. Gisselbrecht, R. Graff, R. Ruppert and H. J. Callot, Inorg. Chem., 2004, 43, 251–263 CrossRef CAS PubMed; (c) K. M. Kadish, M. Lin, E. V. Caemelbecke, G. D. Stefano, C. J. Medforth, D. J. Nurco, N. Y. Nelson, B. Krattinger, C. M. Muzzi, L. Jaquinod, Y. Xu, D. C. Shyr, K. M. Smith and J. A. Shelnutt, Inorg. Chem., 2002, 41, 6673–6687 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Regulatory Body: Environment Protection Agency, USA, Drinking Water Quality Legislation of the United States, 1974, Safe Drinking Water Act PL 93-523, Subchapter 6A of Title 42.
- (a) A. Caballero, R. Martinez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tarranga, P. Molina and J. Vecianaa, J. Am. Chem. Soc., 2005, 127, 15666–15667 CrossRef CAS PubMed; (b) M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414–1418 CrossRef CAS PubMed; (c) W. Lin, L. Yuan, Z. Cao, Y. Feng and L. Long, Chem.–Eur. J., 2009, 15, 5096–5103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation and electrochemical redox data, geometry optimised structures and anion sensing of 1–4. CCDC 1401278 for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra19847j |
| This journal is © The Royal Society of Chemistry 2015 |