
Ruthenium/base-catalyzed ortho-selective C–H arylation of acylarenes with halogenated arylboronates†
Tetsuya Yamamoto‡
and
Tetsu Yamakawa*
Catalysis Group, Sagami Chemical Research Institute, Hayakawa 2743-1, Ayase, Kanagawa 252-1193, Japan. E-mail: t_yamakawa@sagami.or.jp
First published on 3rd December 2015
Abstract
Ruthenium-catalyzed ortho-selective C–H arylation of acylarenes with halogenated aryl-boronates was promoted by the catalytic amount of bases and provided desired halogenated biaryls in excellent yields under 0.2–1 mol% ruthenium catalyst loading.
Introduction
As halogenated 2-acyl-1,1′-biaryls are readily transformed to halogenated fluorene derivatives, they are key intermediates of organic electronic materials such as liquid crystals, organic light-emitting diodes, organic semiconductors, dye sensitized solar cells and conductive polymers.1 Halogenated 2-acyl-1,1′-biaryls are simply synthesized by direct halogenation of corresponding 2-acyl-1,1′-biaryls.2 However, this method has two disadvantages. The one is the limitation of halogenated position and the other is the formation of oligo-halogenated products as by-products. Transition metal-catalyzed cross-coupling such as Suzuki–Miyaura coupling has attracted as a candidate for the practical syntheses of halogenated 2-acyl-1,1′-biaryls.3,4 There are three kinds of the possible combination of substrates in this coupling reaction as depicted in Scheme 1: dihaloarenes and (2-acyl)arylmetallic reagents (i), 2-acyl(halo)haloarenes and aryl-metallic reagents (ii), and (haloaryl)metallic reagents and (2-acyl)haloarenes (iii). In (i) and (ii) where di-haloarenes or 2-acyl(halo)haloarenes are used, the over-reaction inevitably occurs at the second halo group by the use of the general-purpose coupling catalysts. Indeed, most of the reported examples afforded from low to moderate yields of the desired product.4a–c The high yield and selectivity even using 2-acyl-4-bromoiodobenzene was achieved by the Pd-catalyzed Suzuki–Miyaura coupling for (2-acyl-4-bromophenyl)arene synthesis.4d However, the partner is limited to anthrathen-9-ylboronic acid, suggesting that the bulky anthrathen-9-yl group retarded over-reaction. The use of bromochloro or iodochloroarenes is promising to overcome this advantage, because C–Cl bond cannot be activated by the general-purpose coupling catalysts.3 Nevertheless, the product of chloro-substituted biaryls are ones of PCBs derivatives and their syntheses and uses are restricted due to toxicity and carcinogenesis in many countries.5 As well as C–Cl bond, C–S bond also cannot be activated by the general-purpose coupling catalysts. Shi and co-workers reported the Rh-catalyzed cross-coupling of halogenated arylboroxines and 2-acyl(alkylthio)arenes to halogenated 2-acyl-1,1′-biaryls through C–S bond activation.6 However, this reaction afforded low yield of the desired product though it requires the excess amount of aryl-boroxine.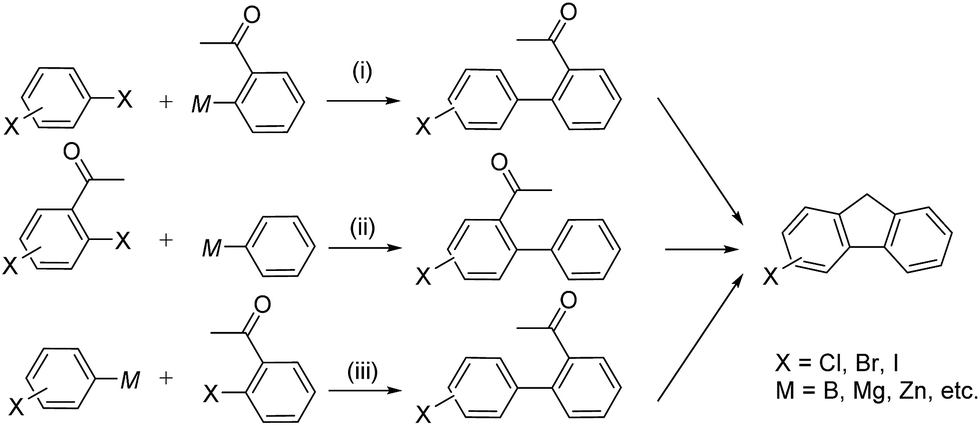 | ||
| Scheme 1 Syntheses of halogenated fluorene derivatives via transition metal-catalyzed cross-coupling. | ||
Thus far, the cross-coupling including C–H bond activation has been also investigated as one of the candidates for halogenated 2-acyl-1,1′-biaryls syntheses. Glorius and co-workers reported the Rh-catalyzed coupling of N,N-diisopropylbenzamides or 2,2-dimethyl-1-phenyl-1-propanone and bromobenzenes to brominated 2-acyl-1,1′-biaryls; diisopropylamide and pivaloyl groups are readily converted to an acyl group by the hydrolysis. In these reactions, both of selective C–H bond activation of N,N-diisopropyl-benzamides or 2,2-dimethyl-1-phenyl-1-propanones at the ortho position of diisopropylamide or pivaloyl group and non-selective C–H activation of bromobenzenes are included, resulting in the mixture of 3′- and 4′-bromo-1,1′-biphenyls.7 Nakamura and co-workers reported Fe-catalyzed arylation of 3-bromo(alkylimino)benzenes with aryl Grignard reagents. In this reaction, only the selective C–H activation occurred at the ortho position of the alkylimino group, providing inevitably 4-bromo-2-alkylimino-1,1′-biphenyl as a sole product.8 Although this reaction is promising for 2-acyl-1,1′-biaryls syntheses through the acid hydrolysis of 2-alkylimino-1,1′-biphenyl, an excess amount of aryl Grignard reagent is necessary.
From the beginning of the 21st century, Ru-catalyzed ortho-selective arylation of arylketones or arylketimines have been developed as one of the most useful synthetic tools for fine chemicals synthesis.9–11 In particular, Kakiuchi C–H arylation has a great potential for bromo- or iodo-substituted 2-acyl-1,1′-biaryls synthesis. This arylation does not include the oxidative addition of aryl halides to ruthenium complex in the catalytic cycle.11 Therefore, the succeeding coupling with aryl boronates or acyl-ketones which would afford multi-arylated products does not occur.
Herein, we examined the Ru-catalyzed ortho-selective C–H arylation of acylarenes using halo-aryl-boronates on the standpoint of the practical use and founds that the addition of catalytic amount of a base and the use of excess amount of acylarenes are the important key to obtain the satisfactory yields of the desired product.
Results and discussion
At first, we examined ruthenium-catalyzed cross-coupling of 1-(naphthalene-1-yl)ethanone 1a and 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (Table 1). In the previous reports of Kakiuchi and co-workers,11 the RuH2(CO)(PPh3)3-catalyzed cross-coupling afforded the corresponding product 3a in low yields with concomitant formation of de-brominated product 4a (entries 1 and 2). The yield of 3a and the selectivity of 3a/4a were improved to 29% and 29/1, respectively, by increasing the used amount of 1a under solvent-free condition (entry 3). Generally, the use of an excess amount of base has activated aryl-boron compounds and promoted a transmetalation of aryl-boron compounds to transition metals.12 Indeed, the cross-coupling also proceeded smoothly in the presence of a stoichiometric amount of Cs2CO3 in this case despite the slight increase in the yield of de-brominated product 4a to 6% (entry 4). Intriguingly, a catalytic amount of Cs2CO3 further promoted the reaction and the yield of de-brominated product was restrained to 2% (entries 5 and 6). Additionally, the 5a/Cs2CO3 catalyzed reaction proceeded smoothly under moderate temperature, 100 °C, in an excellent yield, though the longer reaction time was required (entry 7). The combination of the other ruthenium–triphenylphosphine complexes 5b–d and a catalytic amount of Cs2CO3 similarly showed the satisfactory yield and excellent selectivity of 3a (entries 8–10).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | 1a (eq.) | Cs2CO3 (mol%) | Reaction time (h) | Yield (%) 3ae/4af |
| a Conditions: 1 mmol of 2a was used.b Toluene solvent 1 mL.c Pinacolone solvent 1 mL.d Reaction temperature was 100 °C.e Isolated yield.f GC yield. | |||||
| 1b | 5a (5) | 2.0 | 0 | 2 | 5/1 |
| 2c | 5a (2) | 1.0 | 0 | 2 | 8/2 |
| 3 | 5a (2) | 6.6 | 0 | 2 | 29/1 |
| 4 | 5a (2) | 6.6 | 100 | 1 | 75/6 |
| 5 | 5a (2) | 6.6 | 10 | 1 | 80/2 |
| 6 | 5a (1) | 6.6 | 10 | 1 | 81/2 |
| 7d | 5a (1) | 6.6 | 10 | 12 | 83/3 |
| 8 | 5b (1) | 6.6 | 10 | 1 | 81/3 |
| 9 | 5c (1) | 6.6 | 10 | 12 | 79/4 |
| 10 | 5f (1) | 6.6 | 10 | 12 | 77/2 |

Next, we examined the dependence of the yield of 3a on the used amount of 1a in 0.2 mol% 5b/Cs2CO3 catalyzed cross-coupling of 1a and 2a (Fig. 1). The linear correlation between the yield of 3a and the used amount of 1a was observed in the region of 2–4 equivalent of 1a. The yield was retained by the use of 1a more than 4 equivalent.
 | ||
| Fig. 1 The dependence of the yield of 3a on the used amount of 1a. | ||
Table 2 lists the survey of a base in the cross-coupling of 1a and 2a using 0.2 mol% 5b. Of the bases tested, 20 mol% of Rb2CO3 exhibited the highest turnover number of 435 (entry 2). Virtually the same selectivity of 3a, 95–97% was obtained with any other bases.

On the basis of these results, we synthesized various bromo- or iodo-substituted 2-acyl-1,1′-biaryls using the ruthenium–PPh3 complex 5a or 5b/Cs2CO3 catalyzed reaction (Table 3). 3-Bromo-substituted phenylboronates 2b and 2c reacted smoothly with 1-(naphthalene-1-yl)ethanone 1a as with 4-bromophenyl-boronate 2a to provide corresponding products 3b and 3c in good yields (entries 1 and 2). The reaction of 1a and 2-(4-iodophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2d required lower reaction temperature and longer reaction time than 4-bromophenylboronate 2a for a good yield (entry 3). This catalysis system was not applied to the ortho-substituted phenyl-boronate such as 2-(2-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2e and 2-(4-bromonaphtha-len-1-yl)-5,5-dimethyl-1,3,2-dioxaborinane 2f (entries 4 and 5). With regard to the cyclic alkyl-aryl-ketones, although 2a and α-tetralone 1b afforded 3g in 42% yield, 1-benzosuberone 1c yielded product 3h in a good yield (entries 6 and 7). On the other hand, acyclic acyl-benzenes such as 1-(naphthalene-2-yl)ethan-1-one 1d and 2,2-dimethyl-1-phenylpropan-1-one 1e that have no substituents on ortho-positions were lower reactivity than 1a under this reaction condition (entries 8 and 9). In addition, this catalyst gave 81% yield in the coupling of 1a and 2-phenyl-5,5-dimethyl-1,3,2-dioxaborinane 2g that has no substituent under 0.2 mol% ruthenium catalyst 5b loading (entry 10). In the absence of Cs2CO3, the reaction afforded the coupling product 4a in remarkably low 4% yield (entry 11).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | 1 (eq.) | 2 | Cs2CO3 (mol%) | Reaction temp./time (°C h−1) | 3 yieldb (%) |
| a Conditions: 1 mmol of 2 was used.b Isolated yield.c Detected by GC. | ||||||
| 1 | 5b (0.5) | 1a | 2b | 5 | 150/3 | 3b |
| (6.6) | 80 | |||||
| 2 | 5b (0.5) | 1a | 2c | 10 | 150/3 | 3c |
| (6.6) | 75 | |||||
| 3 | 5b (0.5) | 1a | 2d | 30 | 135/12 | 3d |
| (6.6) | 70 | |||||
| 4 | 5a (2) | 1a | 2e | 20 | 150/3 | 3e |
| (6.6) | Tracec | |||||
| 5 | 5a (2) | 1a | 2f | 20 | 150/3 | 3f |
| (6.6) | Tracec | |||||
| 6 | 5a (1) | 1b | 2a | 20 | 150/2 | 3g |
| (7.5) | 42 | |||||
| 7 | 5a (1) | 1c | 2a | 20 | 150/3 | 3h |
| (6.7) | 74 | |||||
| 8 | 5b (2) | 1d | 2a | 10 | 150/3 | 3i |
| (6.6) | 22 | |||||
| 9 | 5a (2) | 1e | 2a | 20 | 150/3 | 3j |
| (6.0) | Tracec | |||||
| 10 | 5b (0.2) | 1a | 2g | 10 | 150/3 | 4a |
| (6.6) | 81 | |||||
| 11 | 5b (0.2) | 1a | 2g | 0 | 150/3 | 4a |
| (6.6) | 4 | |||||

Generally, aryl-boronic acids are suited for industrial uses, because they are more readily available than aryl-boronates. However, the use of aryl-boronic acid afforded little desired product in the present coupling. Therefore, the one-pot synthesis of 3a from 4-bromophenylboronic acid and 1a in the presence of 2,2-dimethylpropane-1,3-diol was examined in a Dean–Stark apparatus. RuHCl(CO)(PPh3)3/Cs2CO3 catalyst provided 3a in 78% yield in a gram-scale (Scheme 2).
 | ||
| Scheme 2 One-pot synthesis of 3a from 4-bromophenylboronic acid. | ||
A possible catalytic cycle of the C–H arylation with 4-bromophenylboronate 2a and 1-acylnaphthalene 1a is illustrated in Scheme 3. The mechanism depicted has already proposed by Kakiuchi and co-workers.11e,f Thus, catalytically active Ru(0) complex oxidatively inserts in C–H bond at the ortho position of the acyl group. The formed Ru(II) species and 1a provides 1-naphthalenemethoxy Ru(II) complex 6, followed by the transmetalation with 2a to 7. Although the byproduct from 6 and 2a to 7, 5,5-dimethyl-2-[1-(1-naphthyl)ethoxy]-1,3,2-dioxaborinane, could not be isolated presumably because of its un-stability, 2-(1-naphthyl)ethanol that should be originated from this trialkoxyborane was detected by GC. Reductive elimination of the desired product 3a reproduces the catalytically active Ru(0) complex. In this mechanism, a base is not necessary. On the other hand, our C–H arylation with acylarenes and bromo- or iodo-substituted aryl boronates required a base (Table 1). Because an oxidative addition of bromo- or iodo-substituted aryl boronates to Ru(0) complex should occur as a side reaction, catalytically active Ru(0) complex would convert into catalytically inactive bromo- or iodo Ru(II) complexes. As depicted in Scheme 4, Anderson and co-workers previously reported that Ru(0) complex 8 was generated from 5a and readily reacted with 4-bromo- or 4-iodotoluene to (tolyl)ruthenium(II) complex 9 via oxidative addition in moderate yields. Furthermore, thus obtained complex 9 and α-tetralone quickly provided ruthenacycle 10 and toluene selectively via transmetalation through C–H bond cleavage in 99% yield.13
 | ||
| Scheme 3 Catalytic cycle of Ru-catalyzed arylation with 1a and 2a. | ||
 | ||
| Scheme 4 Stoichiometric reaction of Ru(0) complex with 4-bromo- or 4-iodotoluene and α-tetralone.13 | ||
In our case, similar reactions as Scheme 4 will occur. As depicted in Scheme 5, the reaction of Ru(0) complex and boronate 2a to 11 and further transmetalation of 11 and 1a to ruthenacycle 12 will proceeds as a side reaction. Because the transmetalation of ruthenium halide complexes such as 12 between aryl boronate hardly occurs without a base, this side reaction would cause the reduction of the catalytically active Ru(0) complex, resulting in low yield. Since the transmetalation of aryl-boronic acids to Ru(II) complexes is promoted by a base as reported,12 the transmetalation between ruthenacycles 12 and bromophenyl boronate 2a to aryl substituted ruthenacycle 7 would be also accelerated by a base. Thereby, catalytically active Ru(0) complex for Kakiuchi C–H arylation is recovered from 12 in the presence of a base. Likewise, the transmetalation of 6 with 2a in Scheme 3 would be accelerated by a base. Thus, the yield of 3a does not exceed 50% theoretically, if 3a is formed only with the mechanism in Scheme 5. Therefore, the enhancement of the yield from 4% (entry 11) to 81% (entry 10) in Table 3 cannot be explained only by a regeneration of 7 from 12 with a base in Scheme 5.
 | ||
| Scheme 5 Assumed side reaction of Ru(0) with 2a and recovery process of Ru(0). | ||
Moreover, the base could have another contribution in this arylation. As Pignolet and co-workers reported an orthometalation between acetophenone and halogen coordinated Ru(II) complex that has a basic ligand such as acetate ion,14 the orthometalation of acylarenes to halogen coordinated Ru(II) complex such as 5b or 5c would be similarly promoted by a base. Therefore, this orthometalation would generate a ruthenacycle which has the similar structure as ruthenacycle 12 and trigger this catalytic C–H arylation.
Conclusions
In conclusion, we developed the ruthenium/base catalyzed arylation of 2-acylarenes with halogenated aryl-boron compounds to give corresponding halogenated 2-acyl-1,1′-biaryls. The catalytic amount of a base drastically improved the yield and selectivity of the desired product. For example, the turnover number reached more than 435 from 15 in 1-(2-(4-bromophenyl)naphthalene-1-yl)ethanone 3a synthesis by the addition of various bases. The catalytic amount of bases would serve two effects; one is a promotion of the orthometalation of acylarenes by halogen coordinated Ru(II) complexes such as 5b or 5c, the other is an acceleration the transmetalation between ruthenacycles and aryl-boronate.Experimental section
General information
All reactions were carried out under an argon atmosphere. 1H, 13C, and 19F NMR spectra were recorded on a Bruker AVANCE III 400 spectrometer (400.13 MHz) at ambient temperature. The chemical shifts of 1H were reported in delta (δ) units, parts per million (ppm) downfield from tetramethylsilane (0.0 ppm). Chemical shifts of 13C were reported in delta (δ) units, ppm relative to the center of the triplet at 77.0 ppm for CDCl3. The chemical shifts of 19F were referenced to the resonance frequency (0.0 ppm) and 19F 376.27 MHz with a negative sign indicating an upfield shift. High-resolution mass spectra were taken with a JEOL MStaion JMS-700. Low-resolution mass spectra was measured on a Shimadzu GCMS-QP2010. IR spectra were recorded on a HORIBA FT-720. Melting points were recorded on a METTLER TOLEDO MP-70. Commercially available inorganic compounds including ruthenium complexes were used without purification. Freshly-distilled ketones were used in these reactions. Aryl-boronates were prepared from commercially available aryl-boronic acids with 2,2-dimethyl-1,3-propanediol according to the reported procedure.151-(2-(4-Bromophenyl)naphthalene-1-yl)ethan-1-one [3a: Table 2, entry 1]. Using 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (269 mg, 1.00 mmol), cesium carbonate (32.6 mg, 100 μmol), 1-(naphthalene-1-yl)ethan-1-one 1a (1.12 g, 6.6 mmol) and 5b (1.9 mg, 2.0 μmol), the product was obtained in 83% yield (269 mg, 0.827 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 9/1). Mp 103–104 °C; IR (neat, cm−1) 1687, 1591, 1489, 1350, 1209, 1130, 1080, 1011, 864, 839, 818, 791, 756; 1H NMR (400 MHz, CDCl3, ppm): δ 7.83–7.81 (m, 3H), 7.61–7.52 (m, 4H), 7.46 (d, J = 8.4 Hz, 1H), 7.36–7.33 (m, 2H), 2.14 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 207.3, 139.2, 138.5, 134.5, 132.7, 131.9, 131.0, 129.6, 128.8, 128.3, 127.6, 127.0, 126.6, 124.8, 122.5, 32.9; HRMS (FAB) m/z: [M]+ calcd for C18H13BrO: 324.0150. Found: 324.0157.
1-(2-(3-Bromo-5-methylphenyl)naphthalene-1-yl)ethan-1-one [3b: Table 3, entry 1]. Using 2-(3-bromo-5-methylphenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2b (283 mg, 1.00 mmol), cesium carbonate (12.3 mg, 50.0 μmol), 1-(naphthalene-1-yl)ethan-1-one 1a (1.12 g, 6.6 mmol) and 5b (4.8 mg, 10 μmol), the product was obtained in 80% yield (272 mg, 0.802 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 9/1). Mp 105–106 °C; IR (neat, cm−1) 1689, 1597, 1568, 1556, 1350, 1217, 1130, 866, 843, 822, 744, 706, 677; 1H NMR (400 MHz, CDCl3, ppm): δ 7.86–7.83 (m, 3H), 7.58–7.52 (m, 2H), 7.46–7.43 (m, 2H), 7.39 (s, 1H), 7.18 (d, J = 0.6 Hz, 1H), 2.38 (s, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 206.9, 142.2, 140.5, 138.4, 134.5, 132.7, 131.7, 129.5, 129.2, 129.0, 128.8, 128.3, 127.6, 127.1, 126.6, 124.9, 122.6, 32.8, 21.2; HRMS (FAB) m/z: [M]+ calcd for C19H15BrO: 338.0306. Found: 338.0295.
1-(2-(3-Bromo-5-fluorophenyl)naphthalene-1-yl)ethan-1-one [3c: Table 3, entry 2]. Using 2-(3-bromo-5-fluorophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2c (287 mg, 1.00 mmol), cesium carbonate (32.6 mg, 100 μmol), 1-(naphthalene-1-yl)ethan-1-one 1a (1.12 g, 6.6 mmol) and 5b (4.8 mg, 10 μmol), the product was obtained in 75% yield (257 mg, 0.749 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 9/1). Mp 85–87 °C; IR (neat, cm−1) 1682, 1601, 1574, 1427, 1414, 1350, 1217, 1188, 847, 818, 756, 746, 700, 675; 1H NMR (400 MHz, CDCl3, ppm): δ 7.95 (d, J = 8.4 Hz, 1H), 7.93–7.82 (m, 2H), 7.60–7.54 (m, 2H), 7.45–7.42 (m, 2H), 7.31 (ddd, J = 1.9, 2.2, 8.0 Hz, 1H), 7.13 (ddd, J = 1.5, 2.3, 9.0 Hz, 1H), 2.21 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 206.5, 162.4 (d, J = 250.8 Hz), 143.8 (d, J = 8.1 Hz), 138.8, 133.0, 132.9 (d, J = 2.1 Hz), 129.7, 128.7, 128.4, 128.3 (d, J = 2.8 Hz), 127.8, 126.9, 126.7, 124.9, 123.0 (d, J = 9.7 Hz), 118.7 (d, J = 24.3 Hz), 115.6 (d, J = 23.6 Hz), 32.9; 19F NMR (376 MHz, CDCl3, ppm): δ −109.7 (s, 1F); HRMS (FAB) m/z: [M]+ calcd for C18H12BrFO: 342.0056. Found: 342.0049.
1-(2-(4-Iodophenyl)naphthalene-1-yl)ethan-1-one [3d: Table 3, entry 3]. Using 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (316 mg, 1.00 mmol), cesium carbonate (97.8 mg, 300 μmol), 1-(naphthalene-1-yl)ethan-1-one 1a (1.12 g, 6.6 mmol) and 5b (4.8 mg, 5.0 μmol), the product was obtained in 70% yield (259 mg, 0.696 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 9/1). Mp 115–117 °C; IR (neat, cm−1) 1738, 1689, 1485, 1371, 1350, 1211, 1130, 1080, 1005, 970, 953, 862, 837, 816, 789, 754; 1H NMR (400 MHz, CDCl3, ppm): δ 7.93 (d, J = 8.5 Hz, 1H), 7.92–7.82 (m, 4H), 7.58–7.51 (m, 2H), 7.46 (d, J = 8.5 Hz, 1H), 7.23–7.19 (m, 2H), 2.14 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 207.2, 139.8, 138.4, 137.9, 134.6, 132.7, 131.2, 129.6, 128.8, 128.3, 127.6, 127.0, 126.6, 124.8, 94.2, 32.9; HRMS (FAB) m/z: [M]+ calcd for C18H13IO: 372.0011. Found: 372.0019.
8-(4-Bromophenyl)-3,4-dihydronaphthalene-1(2H)-one [3g: Table 3, entry 6]. Using 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (269 mg, 1.00 mmol), cesium carbonate (65.2 mg, 200 μmol), 3,4-dihydronaphthalene-1(2H)-one 1b (1.10 g, 7.5 mmol) and 5a (9.2 mg, 10 μmol), the product was obtained in 42% yield (125 mg, 0.415 mmol) as a pale yellow solid after column chromatography (hexane/ethyl acetate: 10/0 to 8/1). Mp 109–111 °C; IR (neat, cm−1) 2935, 2858, 1680, 1583, 1489, 1458, 1269, 1250, 1207, 1178, 1068, 1022, 1009, 930, 822, 796, 766, 684; 1H NMR (400 MHz, CDCl3, ppm): δ 7.50–7.46 (m, 2H), 7.42 (dd, J = 7.6, 7.6 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.09–7.06 (m, 3H), 3.01 (t, J = 6.1 Hz, 2H), 2.61 (t, J = 6.6 Hz, 2H), 2.19–2.09 (m, 2H); 13C NMR (100 MHz, CDCl3, ppm): δ 198.4, 145.8, 142.7, 141.9, 132.0, 131.0, 130.9, 130.1, 129.9, 128.6, 120.8, 40.5, 30.7, 23.0; HRMS (FAB) m/z: [M]+ calcd for C16H13BrO: 300.0150. Found: 300.0155.
4-(4-Bromophenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one [3h: Table 3, entry 7]. Using 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (269 mg, 1.00 mmol), cesium carbonate (65.2 mg, 200 μmol), 6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one 1c (1.07 g, 6.7 mmol) and 5a (9.2 mg, 20 μmol), the product was obtained in 74% yield (233 mg, 0.740 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 8/1). Mp 151–152 °C; IR (neat, cm−1) 2927, 2860, 1684, 1587, 1454, 1242, 1178, 1103, 1078, 1005, 876, 837, 823, 796, 775, 758, 688; 1H NMR (400 MHz, CDCl3, ppm): δ 7.49–7.46 (m, 2H), 7.36 (dd, J = 7.6, 7.6 Hz, 1H), 7.20 (dd, J = 1.1, 7.6 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 7.12–7.08 (m, 2H), 2.81 (t, J = 6.3 Hz, 2H), 2.66–2.63 (m, 2H), 1.95–1.84 (m, 4H); 13C NMR (100 MHz, CDCl3, ppm): δ 209.9, 140.3, 139.9, 139.0, 137.9, 131.4, 130.3, 130.0, 128.7, 128.2, 121.4, 42.8, 32.5, 25.6, 22.8; HRMS (FAB) m/z: [M]+ calcd for C17H15BrO: 314.0306. Found: 314.0312.
1-(2-(4-Bromophenyl)naphthalene-1-yl)ethan-1-one [3i: Table 3, entry 8]. Using 2-(4-bromophenyl)-5,5-dimethyl-1,3,2-dioxaborinane 2a (269 mg, 1.00 mmol), cesium carbonate (32.6 mg, 100 μmol), 1-(naphthalene-2-yl)ethan-1-one 1d (1.13 g, 6.6 mmol) and 5b (19.2 mg, 20.0 μmol), the product was obtained in 22% yield (70 mg, 0.215 mmol) as a white solid after column chromatography (hexane/ethyl acetate: 10/0 to 10/1). Mp 83–85 °C; IR (neat, cm−1) 1689, 1489, 1458, 1440, 1419, 1392, 1360, 1279, 1269, 1244, 1209, 1191, 1130, 1072, 1009, 982, 964, 951, 914, 899, 823, 761, 667; 1H NMR (400 MHz, CDCl3, ppm): δ 8.11 (s, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.80 (s, 1H), 7.62–7.55 (m, 4H), 7.30 (d, J = 8.3 Hz, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 203.3, 140.0, 138.6, 136.2, 134.0, 131.8, 130.5, 129.6, 128.9, 128.7, 128.3, 127.8, 127.1, 122.0, 30.4; HRMS (ESI orbitrap) m/z: [M + Na]+ calcd for C18H13BrONa: 347.0042. Found: 347.0036.
1-(2-Phenylnaphthalene-1-yl)ethan-1-one [4a: Table 3, entry 10]11f. Using 2-phenyl-5,5-dimethyl-1,3,2-dioxaborinane 2f (190 mg, 1.00 mmol), cesium carbonate (32.6 mg, 100 μmol) and 5b (1.9 mg, 2.0 μmol), the product was obtained in 81% yield (199 mg, 0.809 mmol) as a pale yellow liquid after column chromatography (hexane/ethyl acetate: 10/0 to 9/1). 1H NMR (400 MHz, CDCl3, ppm): δ 7.94 (d, J = 8.4 Hz, 1H), 7.92–7.85 (m, 2H), 7.58–7.39 (m, 8H), 2.09 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 207.4, 140.3, 138.3, 136.0, 132.6, 129.4, 129.4, 128.8, 128.7, 128.2, 128.0, 127.4, 126.3, 124.8, 32.7; GCMS (EI): 246.
Acknowledgements
We thank the Instrumental Analysis Laboratory, University of Toyama, and Prof. Mino, Chiba University, for the HRMS measurements. Technical assistance for the additional experiment by Mr Ryo Akiyama (undergraduate student of Tokyo Denki University) is gratefully acknowledged.Notes and references
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS PubMed.
- Selected examples of a bromination of biaryls: (a) B. M. Schmidt, B. Topolinsky, M. Yamada, S. Higashibayashi, M. Shionoya, H. Sakurai and D. Lentz, Chem.–Eur. J., 2013, 19, 13872 CrossRef CAS PubMed; (b) S. Song, S. Park, S. Kwon, B. H. Lee, J. Y. Shim, J. Lee, S. H. Park, Y. Jin, I. Kim, K. Lee and H. Suh, Sol. Energy Mater. Sol. Cells, 2012, 105, 229 CrossRef; (c) M.-S. Gong, H.-S. Lee and Y.-M. Jeon, J. Mater. Chem., 2010, 20, 10735 RSC.
- For selected recent reviews: (a) R. Rossi, F. Bellina and M. Lessia, Adv. Synth. Catal., 2012, 354, 1181 CrossRef CAS; (b) R. Rossi, F. Bellina and M. Lessi, Tetrahedron, 2011, 67, 6969 CrossRef CAS.
- Brominated 2-acyl-1,1′-biaryls synthesis via Suzuki-Miyaura coupling: (a) M. Nawaz, I. Ullah, O.-U.-R. Abid, A. Villinger and P. Langer, Eur. J. Org. Chem., 2011, 6670 CrossRef CAS; (b) Y. Wu, X. Hao, J. Wu, J. Jin and X. Ba, Macromolecules, 2010, 43, 731 CrossRef CAS; (c) L. Lunazzi, A. Mazzanti and M. Minzoni, J. Org. Chem., 2007, 72, 2501 CrossRef CAS PubMed; (d) C. Yang, J. Jacob and K. Müllen, Macromolecules, 2006, 39, 5696 CrossRef CAS.
- For selected examples: (a) Q. Zhang, M. Lu, C. Wang, J. Du, P. Zhou and M. Zhao, Environ. Pollut., 2014, 189, 169 CrossRef CAS; (b) M. V. Berghe, L. Weijs, S. Habran, K. Das, C. Bugli, S. Pillet, J.-F. Rees, P. Pomeroy, A. Covaci and C. Debier, Environ. Res., 2013, 120, 18 CrossRef; (c) M. P. Ward, C. Jablonski, B. Semel and D. Soucek, Ecotoxicology, 2010, 19, 1513 CrossRef CAS PubMed; (d) S. Akzinnay, F. Bisaro and C. S. Cazin, Chem. Commun., 2009, 5752 RSC; (e) D. G. Patterson Jr, W. E. Turner, S. P. Caudill and L. L. Needham, Chemosphere, 2008, 73, 261 CrossRef PubMed; (f) S. H. Safe, Crit. Rev. Toxicol., 1994, 24, 87 CrossRef CAS PubMed.
- F. Pan, H. Wang, P.-X. Shen, J. Zhaob and Z.-J. Shi, Chem. Sci., 2013, 4, 1573 RSC.
- J. Wencel-Delord, C. Nimphius, F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 2247 CrossRef CAS.
- (a) L. Ilies, M. Kobayashi, A. Matsumoto, N. Yoshikai and E. Nakamura, Adv. Synth. Catal., 2012, 354, 593 CrossRef CAS; (b) N. Yoshikai, S. Asako, T. Yamakawa, L. Ilies and E. Nakamura, Chem.–Asian J., 2011, 6, 3059 CrossRef CAS; (c) N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 2925 CrossRef CAS PubMed.
- For selected recent reviews: (a) L. Ackermann, Org. Process Res. Dev., 2015, 18, 260 CrossRef; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211 CrossRef CAS PubMed.
- (a) E. Diers, N. Y. P. Kumar, T. Mejuch, I. Marek and L. Ackermann, Tetrahedron, 2013, 69, 4445 CrossRef CAS; (b) B. Li, K. Devaraj, C. Darcel and P. H. Dixneuf, Tetrahedron, 2012, 68, 5179 CrossRef CAS; (c) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221 CrossRef CAS.
- (a) K. Kitazawa, T. Kochi, M. Nitani, Y. Ie, Y. Aso and F. Kakiuchi, Chem. Lett., 2011, 40, 300 CrossRef CAS; (b) S. Hiroshima, D. Matsumura, T. Kochi and F. Kakiuchi, Org. Lett., 2010, 12, 5318 CrossRef CAS PubMed; (c) K. Kitazawa, M. Kotani, T. Kochi, M. Langeloth and F. Kakiuchi, J. Organomet. Chem., 2010, 695, 1163 CrossRef CAS; (d) K. Kitazawa, T. Kochi, M. Sato and F. Kakiuchi, Org. Lett., 2009, 11, 1951 CrossRef CAS PubMed; (e) F. Kakiuchi, Y. Matsuura, S. Kan and N. Chatani, J. Am. Chem. Soc., 2005, 127, 5936 CrossRef CAS; (f) F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698 CrossRef CAS PubMed.
- (a) K. Li, N. Hu, R. Luo, W. Yuan and W. Tang, J. Org. Chem., 2013, 78, 6350 CrossRef CAS PubMed; (b) Y. Yamamoto, K. Kurihara, Y. Takahashi and N. Miyaura, Molecules, 2013, 18, 14 CrossRef CAS PubMed; (c) Y. Yamamoto, T. Shirai and N. Miyaura, Chem. Commun., 2012, 48, 2803 RSC; (d) M. Kawatsura, K. Kamesaki, M. Yamamoto, S. Hayase and T. Itoh, Chem. Lett., 2010, 39, 1050 CrossRef CAS; (e) Y. Yamamoto, K. Kurihara and N. Miyaura, Angew. Chem., Int. Ed., 2009, 48, 4414 CrossRef CAS PubMed; (f) Y. Na, S. Park, S. B. Han, H. Han, S. Ko and S. Chang, J. Am. Chem. Soc., 2004, 126, 250 CrossRef CAS PubMed; (g) H. Kondo, N. Akiba, T. Kochi and F. Kakiuchi, Angew. Chem., Int. Ed., 2015, 54, 9293 CrossRef CAS PubMed.
- H. Grounds, J. C. Anderson, B. Hayter and A. J. Blake, Organometallics, 2009, 28, 5289 CrossRef CAS.
- M. F. McGuiggan and L. H. Pignolet, Inorg. Chem., 1982, 21, 2523 CrossRef CAS.
- (a) J. Takaya, S. Tadami, K. Ukai and N. Iwasawa, Org. Lett., 2008, 10, 2697 CrossRef CAS PubMed; (b) S. L. Zheng, S. Reid, N. Lin and B. H. Wang, Tetrahedron Lett., 2006, 47, 2331 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR and 19F NMR spectra. See DOI: 10.1039/c5ra19810k |
| ‡ Present address: Department of green and sustainable chemistry, Tokyo Denki University, Senju-Asahi-cho 5, Adachi-ku, Tokyo 120-8551, Japan. E-mail: E-mail: t-yamamoto@mail.dendai.ac.jp |
| This journal is © The Royal Society of Chemistry 2015 |