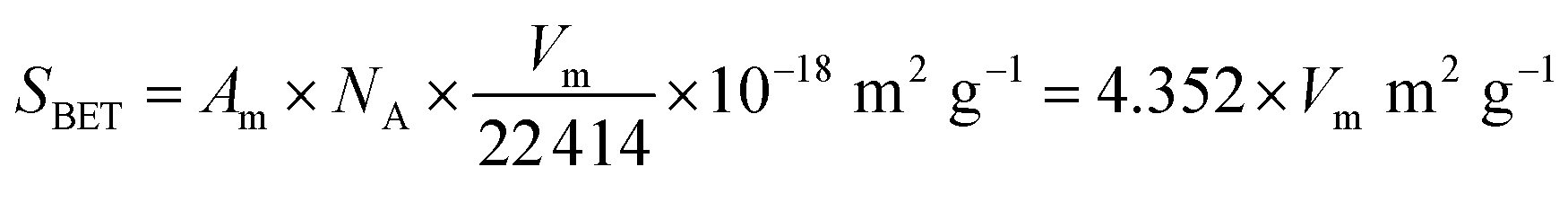DOI:
10.1039/C5RA19764C
(Paper)
RSC Adv., 2016,
6, 5611-5620
Novel Al2O3–SiO2 composite aerogels with high specific surface area at elevated temperatures with different alumina/silica molar ratios prepared by a non-alkoxide sol–gel method†
Received
24th September 2015
, Accepted 22nd December 2015
First published on 23rd December 2015
Abstract
We have developed a new sol–gel route to synthesise Al2O3–SiO2 composite aerogels with different alumina/silica (Al/Si) molar ratios using an inexpensive inorganic salt. The approach is straightforward, inexpensive, and it produces monolithic mesoporous material with high specific surface area heat-treated at elevated temperatures. The effects of different Al/Si molar ratios and calcination temperatures on the microstructures and properties of Al2O3–SiO2 composite aerogels are investigated in this study. Results show that SiO2 is essentially amorphous, while Al2O3 predominately exists as polycrystalline boehmite for the as-dried composite aerogels. With the increase of Al/Si molar ratios, the morphologies change from connected spheroidal particles to nanometer-sized fibrous particles and web-like microstructures with varying diameters. As the heat treatment temperature increases to 600 °C, structural transition from boehmite to γ-Al2O3 occurs within all the composite aerogels, and mullitization firstly occurs with the Al/Si molar ratio of 1 at around 1000 °C. The specific surface area undergoes an increase–decrease–increase process at 600 °C and 1200 °C for the composite aerogels with different Al/Si molar ratios. The specific surface area is as high as 166 m2 g−1 at 1200 °C for the sample with an Al/Si molar ratio of 8, which is higher than ever reported. The thermal conductivities of mullite fiber mat reinforced aerogel composites at room temperatures are 0.023 W m−1 K−1, 0.029 W m−1 K−1 and 0.025 W m−1 K−1 with the Al/Si molar ratios of 2, 3 and 8, respectively, suitable for efficient thermal insulations uses.
1 Introduction
Alumina aerogels are nano-porous materials with low density, high porosity and high surface area. Due to their unique nanostructure, they show relatively high strength, enhanced thermal stability, high specific surface area and chemical stabilities.1,2 Thus, they have drawn great interests in a wide range of application prospects in advanced aircraft, spacecraft and catalyst carriers at high temperatures.3 However, practical applications of alumina aerogels have been restricted due their limited heat resistance and the drastic decrease of specific surface area upon heat treatment at temperatures over 1000 °C due to phase transitions of alumina nanoparticles. Thus, significant efforts have been dedicated to thermal stability improvement of alumina aerogels via maintaining the nanostructures and specific surface areas of alumina aerogels at elevated temperatures.4,5 One way to solve the problem is to synthesise alumina aerogel through modification in the preparation process. Zu1 et al. reported a method of preparing Al2O3 aerogel which can be used at temperatures as high as 1300 °C via a novel acetone–aniline in situ water formation (ISWF) method combined with supercritical fluid modification (SCFM) and hexamethyldisilazane gas phase medication. Wang6 et al. developed a method of preparing monolithic super heat-resistant trimethylethoxysilane-modified alumina aerogels based on the ISWF and SCFM techniques. The resulting alumina aerogel shrank little and possessed a high specific surface area of 147 m2 g−1 after heat treatment of 1200 °C for 2 h. The thermal stability of alumina aerogels was much improved, however, the method was a little complex, expensive and the thermal conductivity was a little higher than the conventional silica aerogels. Another two methods were commonly reported for the suppression of alumina phase transition: one was the addition of other elements, such as Si, La, Ba, or Zr,4,7 and the other was to decrease the bulk density of the alumina powders.8 Thus many researchers have paid attention to Al2O3–SiO2 composite aerogel with much low bulk density and silica-doped effect in order to obtain composite with higher heat resistance and larger specific surface area at elevated temperatures.9 Lin10 et al. proposed a method using aluminum sec-butoxide as alumina source and tetraethoxysilane as silicon source to prepare SiC-coated mullite fiber reinforced Al2O3–SiO2 aerogel composite. The composite possessed heat resistance at temperatures over 1200 °C since the pyrolysis was conducted at such a high temperature, and the thermal conductivity of the composite was as low as 0.049 W m−1 K−1 at 1000 °C. Feng11 et al., for example, have shown the effect of the silica content on the structures and properties of the Al2O3–SiO2 aerogel. They had showed that when the silica content is 6.1–13.1 wt%, Al2O3–SiO2 composite aerogel possessed a larger specific surface area (339–445 m2 g−1) than the pure alumina aerogel (157 m2 g−1) at 1000 °C, as a result of the suppression of the phase transition. Osaki12,13 et al. proposed a method using aluminum isopropoxide as alumina source and tetraethyl orthosilicate as silicon source to prepare Al2O3–SiO2 aerogel. The addition of silica particles surrounding the γ-Al2O3 inhibited the contacts of Al2O3 particles, thus decreasing the probability of crystal nucleation, and α-Al2O3 phase transition was largely inhibited. It possessed a large specific surface area of 47 m2 g−1 at 1200 °C. Although preparation of alumina aerogel monoliths from aluminum alkoxide has been used to prepare a variety of alumina aerogels with large specific surface area,8,14,15 this method was typically difficult due to complex chemical pathways leading to gelation, the susceptibility to cracking during drying and especially the high reactivity of the aluminum alkoxide, which easily causes precipitation.16 Hurwitz17 et al. directly used boehmite powder as alumina source to prepare Al2O3–SiO2 composite aerogel, the specific surface area was as high as 266 m2 g−1 at 1100 °C, whereas the phase transition of γ-Al2O3/η-Al2O3 happened as early as 600 °C. On the other hand, the boehmite powder required to be dispersed in 0.09 M nitric acid solution or in water and sonicated using ultrasonic processor, which was also a complex procedure. Thus, an attempt to develop a sol–gel method that utilizes simple aluminum salts in the preparation of high-surface-area Al2O3–SiO2 composite aerogel, avoiding the use of highly reactive alkoxide precursors, is worthwhile.
The objective of this work is to develop a new convenient sol–gel method that facilitates the fabrication of Al2O3–SiO2 composite aerogel, which possesses high specific surface area at elevated temperatures and low thermal conductivity at room temperature. Conventionally, the silicon sol and the aluminum sol has to be prepared separately followed by mixing in one pot, since the hydrolysis rate of them is much different. Herein in our work, the monolithic Al2O3–SiO2 composite aerogels with different Al/Si molar ratios were prepared by simply mixing the reactants in one pot, followed by gel, aging, solvent exchange and C2H5OH supercritical drying. Al2O3–SiO2 sols were synthesized by using cheap inexpensive inorganic salt (AlCl3·6H2O) and tetraethoxysilane (TEOS) as precursors, ethanol (EtOH) as solvent, deionized water (H2O) as hydrolyzing agent, propylene oxide (PO) as network forming agent, and no catalyst was used in the total procedure. Details of synthesis and discussions of the effects of Al/Si molar ratios and calcination temperatures on the microstructures and properties of Al2O3–SiO2 composite aerogels are given below.
2 Experimental procedures
2.1 Synthesis
AlCl3·6H2O, TEOS, EtOH, H2O and PO were used as raw materials. All of the reagents and solvents were analytical grade and used as received without further purification. Al2O3–SiO2 sols were prepared according to the following steps. AlCl3·6H2O, C2H5OH, H2O, TEOS were directly mixed in a pot with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:50:(0.125, 0.25, 0.33, 0.5, 1), stirring for about 90 min at 50 °C for complete hydrolysis and then cooled to room temperature. Subsequently, desired amounts of PO (molar ratio PO/Al = 8) was slowly dropped into the clear solution (propylene oxide was transferred by syringe through a septum, so as to reduce laboratory exposure and ensure safety. It is listed by the US Environmental Protection Agency as a group B2 possible human carcinogen, EPA Bulletin 75-96-9, crated April 1992, revised January 2000). The reaction mixture was further stirred for 10 min at room temperature, transferred to plastic molds, and the solutions were allowed to gel at room temperature within 3 h. The wet gels were aged at room temperature for 2 d and subsequently the gel parts were soaked in a bath of absolute ethanol for 5 d to exchange the water and reaction byproducts from the pores of the materials. After aging and solvent exchange, the wet gels were placed in an autoclave with volume of 0.5 L, after which the temperature was ramped up to 270 °C with the heating rate of 1 °C min−1 while maintaining a pressure of 100 bar for 2 h. The autoclave was then depressed at a rate of approximately 20 bar h−1. The autoclave vessel was evacuated after decompression for about 200 min, thus affording the Al2O3–SiO2 composite aerogels with different Al/Si molar ratios. In order to identify the microstructures and properties of the Al2O3–SiO2 composite aerogels at elevated temperatures, they were all heat-treated with the same heating rate of 3 °C min−1 to the peak temperatures and maintained at that level for 2 h in a muffle furnace. It was noted here that the sample heat-treated at 600 °C with Al/Si molar ratio of 8 was denoted as AS = 8–600 in this article.
:15:50:(0.125, 0.25, 0.33, 0.5, 1), stirring for about 90 min at 50 °C for complete hydrolysis and then cooled to room temperature. Subsequently, desired amounts of PO (molar ratio PO/Al = 8) was slowly dropped into the clear solution (propylene oxide was transferred by syringe through a septum, so as to reduce laboratory exposure and ensure safety. It is listed by the US Environmental Protection Agency as a group B2 possible human carcinogen, EPA Bulletin 75-96-9, crated April 1992, revised January 2000). The reaction mixture was further stirred for 10 min at room temperature, transferred to plastic molds, and the solutions were allowed to gel at room temperature within 3 h. The wet gels were aged at room temperature for 2 d and subsequently the gel parts were soaked in a bath of absolute ethanol for 5 d to exchange the water and reaction byproducts from the pores of the materials. After aging and solvent exchange, the wet gels were placed in an autoclave with volume of 0.5 L, after which the temperature was ramped up to 270 °C with the heating rate of 1 °C min−1 while maintaining a pressure of 100 bar for 2 h. The autoclave was then depressed at a rate of approximately 20 bar h−1. The autoclave vessel was evacuated after decompression for about 200 min, thus affording the Al2O3–SiO2 composite aerogels with different Al/Si molar ratios. In order to identify the microstructures and properties of the Al2O3–SiO2 composite aerogels at elevated temperatures, they were all heat-treated with the same heating rate of 3 °C min−1 to the peak temperatures and maintained at that level for 2 h in a muffle furnace. It was noted here that the sample heat-treated at 600 °C with Al/Si molar ratio of 8 was denoted as AS = 8–600 in this article.
2.2 Characterizations
Thermal gravimetric analysis (TGA) and differential scanning calorimetry analysis (DSC) were performed by NETZSCH STA449C thermogravimetric analyzer under a constant nitrogen flow of 30 mL min−1 at a heating rate of 10 °C min−1 to 1200 °C. X-ray diffraction (XRD) patterns were recorded using a Rigaku Smart Lab 3000, a diffractometer with CuKα1 radiation (λ = 0.15406 nm). The X-ray tube was operated at 35 kV and 30 mA. Scanning electron microscopy (SEM) was conducted using a LEO-1530VP field emission scanning electron microscope. Photographs of the aerogels were taken with a transmission electron microscope (TEM) using JEOL JEM-2100 (UHR) operating at the acceleration voltage of 200 kV. Surface areas, pore volume and pore distribution were measured by nitrogen adsorption/desorption porosimetry by using a Micromeritics ASAP2020 surface area and pore distribution analyzer after the samples were degassed in a vacuum at 300 °C for 5 hours. The thermal conductivities of the mullite fiber mat reinforced Al2O3–SiO2 composite aerogels (40 mm × 40 mm × 10 mm) were investigated by the measuring apparatus (TPS2500, Hot Disk, Sweden) using a transient plane heat source method at room temperature of 25 °C.
3 Results and discussion
Fig. 1 exhibits the photos of wet gels and aerogels with different appearances, and some experimental values including the masses, volumes and bulk densities of the composite aerogels with different molar ratios are presented in Table S1.† Fig. 2 shows the schematic representation of growth mechanism of Al2O3–SiO2 composite aerogel via C2H5OH supercritical drying.18 The theoretical density can be calculated as the following equation:| |
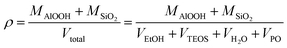 | (1) |
where MAlOOH, MSiO2 are the masses of boehmite and SiO2 aerogel part and VEtOH, VTEOS, VH2O, VPO are the volumes of EtOH, TEOS, H2O and PO added to the solution in the total procedure. The as-prepared Al2O3–SiO2 composite aerogels exhibit crack-free and translucent monolith in appearance. The bulk densities of the five samples (AS = 8, 4, 3, 2, 1) are 0.053 g cm−3, 0.064 g cm−3, 0.076 g cm−3, 0.085 g cm−3 and 0.096 g cm−3, respectively, which are below 0.1 g cm−3, comparable to aluminosilicate aerogel, as reported by Hurwitz et al.17 It is reasonable that the bulk densities increase with a decrease in Al/Si molar ratios s due to the increase of MSiO2 in eqn (1). During the preparation, the PO is employed as the proton scavenger and gel promoter. It can slowly consume the protons in an acidic condition and mildly facilitate the condensation mildly, which results in the more interconnected network.19 The sol–gel mechanism can be described as eqn (2)–(4):20–23
 |
| | Fig. 1 Photographs of Al2O3–SiO2 composite wet gel (a and b) and aerogel (c and d) with different shapes. | |
 |
| | Fig. 2 Schematic representation of growth mechanism of Al2O3–SiO2 composite aerogel (AS = 1). | |
(1) Hydrolysis:
| |
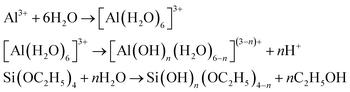 | (2) |
| |
 | (3) |
(2) Condensation:
| |
 | (4) |
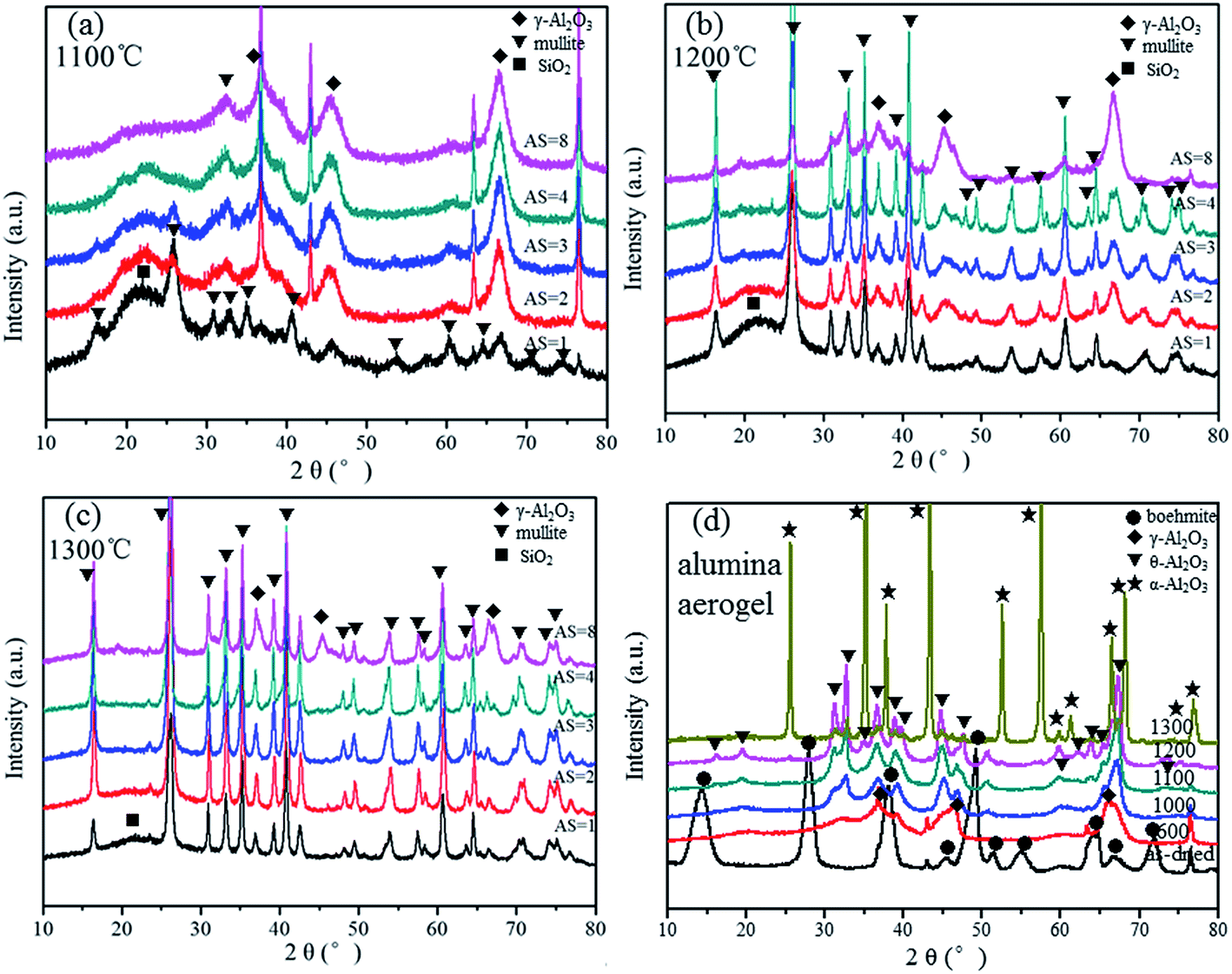
Fig. 3 provides the XRD patterns of Al2O3–SiO2 composite aerogel with different Al/Si molar ratios, which are heat-treated at different temperatures. For the as-dried samples, with an increase of Al/Si molar ratios, the broad diffraction peaks at 15°, 28°, 38°, 49°, 56°, 65° and 72°, which corresponds to pseudoboehmite (AlO(OH), PDF no. 83-2384), become more evident. It exists as polycrystalline boehmite instead of an amorphous morphology.20 After heat treatment at 600 °C for 2 h, the γ-Al2O3 (PDF no. 1-1308) pattern begins to emerge in the composite aerogel. The difference is that the peaks of γ-Al2O3 become stronger with the increase in Al/Si molar ratios, and boehmite still exists in the composite for AS = 1, 2 and 3. Changes are not observed after additional heat treatment at 800 °C for 2 h. With a higher heat treatment temperature at 1000 °C, the mullite phase (PDF no. 79-1451) begins to appear in the composite with AS = 1, whereas there are only the γ-Al2O3 phase and SiO2 phase for the other samples. Conventionally, the mullite phase is formed as described by the chemical eqn (5) as follows:24,25
| | |
3Al2O3 + 2SiO2 → 3Al2O3·2SiO2(mullite)
| (5) |
 |
| | Fig. 3 XRD patterns of Al2O3–SiO2 composite aerogel with different Al/Si molar ratios heat-treated at different temperatures (a) as-dried, (b) 600 °C, (c) 800 °C and (d) 1000 °C. Note that the sharp peaks at 37°, 43°, 63° and 76° are due to the aluminum sample holder. | |
The stoichiometric Al/Si molar ratio should be 3:1, which seems inconsistent with the experimental result. However, since the reactant are mixed in one pot, there exits three different kinds of Al–Si net sketches as Fig. S1.† When the Al/Si molar ratio is larger than 1, rich-SiO2 net and rich-Al2O3 net part will exist in the composite at the same time, and they will form SiO2 phase and Al2O3 phase, separately. In other words, when the Al/Si molar ratio is 1, it forms a monophasic gel, whereas diphasic gel forms when the molar ratio is larger than 1. It is demonstrated by the XRD patterns which contains SiO2 and the γ-Al2O3 phase. Thus, the mullitization process is inhibited with a larger Al/Si molar ratio. As is shown in Fig. 4, the mullite phase becomes more apparent at 1100 °C for AS = 1, and it begins to appear in the other samples with larger Al/Si molar ratios. However, γ-Al2O3 exists in the composite as the main phase since the peaks of γ-Al2O3 are so strong with large Al/Si molar ratios, especially for the AS = 8 at temperature as high as 1100 °C. After heat treatment at 1200 °C, all the samples give more narrow and intense peaks of the mullite except the sample of AS = 8. The peaks corresponding to γ-Al2O3 still evidently remains in the composite. After further heat treatment at 1300 °C for 2 h, all the samples exhibit intense peaks of the mullite, whereas the sample of AS = 8 still possesses small amount of γ-Al2O3. For comparison, Fig. 4(d) shows the XRD patterns of pure Al2O3 aerogel heat-treated at different temperatures, which is also derived from the PO method using AlCl3·6H2O as the alumina source. It is worth noting that with the increase of heat treatment temperature, the Al2O3 aerogel follows the sequence boehmite → γ-Al2O3 → θ-Al2O3 → α-Al2O3. The results infer that α-Al2O3 phase transition is inhibited during heat treatment because γ-Al2O3 stably exists at temperature as high as 1300 °C for the sample of AS = 8. This phenomenon is favorable to homogeneous nanostructures and low thermal conductivity, since α-Al2O3 phase transition (hexagonal close-packed structure) causes volume shrinkage and sintering, compared with other Al2O3 crystal forms (spinel structure), which decreases the heat insulation performance of Al2O3–SiO2 composite aerogel.
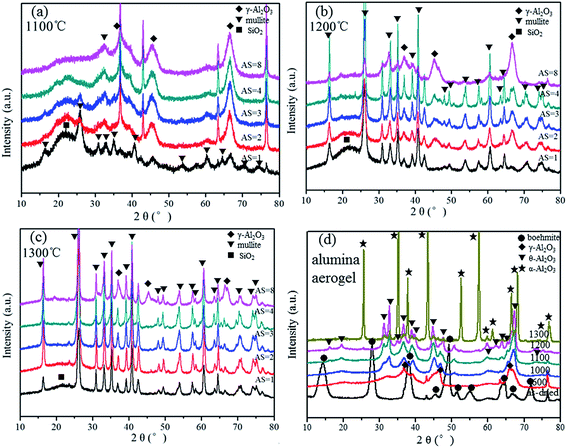 |
| | Fig. 4 XRD patterns of Al2O3–SiO2 composite aerogel with different Al/Si molar ratios heat-treated at different temperatures (a) 1100 °C, (b) 1200 °C, (c) 1300 °C, combined with (d) alumina aerogel. | |
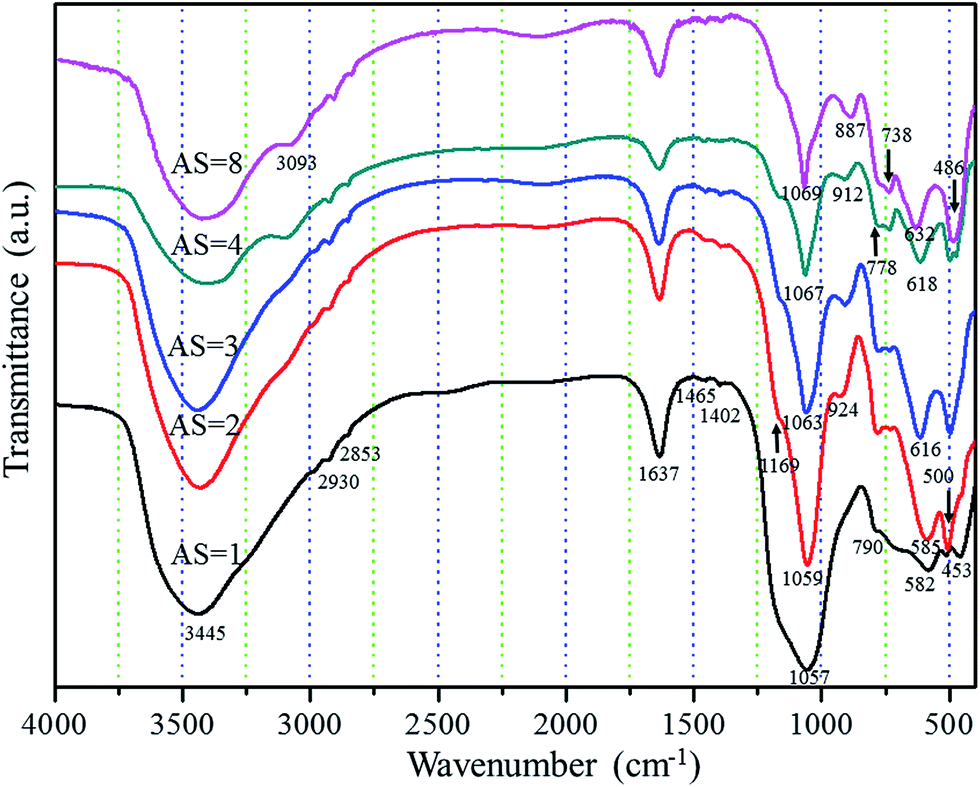
Fig. 5 presents the FTIR analysis of the as-dried samples with different Al/Si molar ratios. The characteristic peaks of the composite aerogels draw close to pure Al2O3 aerogel with the increase of Al/Si molar ratios. The presence of some water molecules is evidenced by the bands at 3445 cm−1 and 1637 cm−1. These two bands do not show significant changes with the decrease of Al/Si molar ratios. The presence of a small amount of organic residues is evidenced by the typical IR bands at 2981 cm−1, 2930 cm−1, 2853 cm−1, 1465 cm−1 and 1402 cm−1, which is caused by C–H vibration of hydrocarbon groups.26 It is well known that the lamellar structure of AlOOH (Fig. S2†) has been previously reported by Yarbrough and Roy.27 The OH groups within the structure could form zigzag chains between the planes of oxygen ions, which could lead to the above two distinct OH stretching modes due to their crystallographically inequivalent coupling effect.28 The band at 1069 cm−1 is attributed to the νs Al–O–H modes of boehmite, whereas the band at 3092 cm−1 is assigned to the νas Al–O–H stretching vibration of boehmite.28 The bands at 778 cm−1, 632 cm−1, 887 cm−1 and 486 cm−1 are attributed to the Al–O mode of boehmite, which is similar to the alumina aerogel reported by Zu3 et al. It is worth noting that the bands at 1069 cm−1 and 632 cm−1 corresponding to boehmite shift towards lower wavenumbers with the decrease of Al/Si molar ratios, which is caused by the presence of Si in its environment. This phenomenon has been observed and confirmed more than once.29 The peaks appeared at near 1169 cm−1 and 738 cm−1 are important signs to declare the existence of hetero-linkage Si–O–Al band in the sample, which is favorable to mullite formation in the further reaction. The above FTIR analysis is consistent with the XRD results.
 |
| | Fig. 5 FTIR spectra of the as-dried Al2O3–SiO2 composite aerogels with different Al/Si molar ratios. | |
Fig. 6 presents the TG and DSC curves of the as-dried Al2O3–SiO2 composite aerogels with different Al/Si molar ratios heat-treated to 1200 °C in flowing argon. The thermogram profile can be divided into three main regions: (1) 20–200 °C, (2) 200–500 °C, and (3) 500–1200 °C. The first region is caused by the evolution of physically adsorbed ethanol and water, whereas the second region is attributed to the continuous structural transition from boehmite to γ-Al2O3 as follows:28,30,31
| | |
2AlO(OH) → γ-Al2O3 + H2O
| (6) |
 |
| | Fig. 6 TG and DSC curves of as-dried Al2O3–SiO2 composite aerogel with different Al/Si molar ratios under flowing argon. | |
The theoretical weight loss of 15% can be calculated with this the equation, which is a little larger than the practical weight loss (for AS = 8, the value is 13.9%) in this region because there is little amount of amorphous SiO2, which doesn't lose weight in the heat treatment process. When the SiO2 content of the composite aerogel increase, weight loss decreases during the second region. It is found that a broad endothermic peak appears in the second region, which is caused by the removal of hydroxyl in the skeleton of Si–OH and Al–OH, followed by the structural transition as eqn (6). There is almost no weight loss in the third region, whereas there is a sharp but small exothermic peak in the DSC curve, which is attributed to the mullitization as eqn (5) at elevated temperatures. It is worth noted that with the increase of Al/Si molar ratios, the mullitization temperature increases as well. This is consistent with the XRD patterns because it also indicates that the mullite phase begins to appear at a temperature of 1000 °C for the sample of AS = 1, and mullitization temperature is as high as 1100 °C for the sample of AS = 8. The production of mullite phase is beneficial for improving the properties of the composite aerogel due to its various attractive properties which include high thermal stability, a high melting point and chemical stability.32,33 The formation temperature in this experiment is lower than the temperature required in the solid state (such as powder mixture) method, in which the temperature ranges from 1240 °C to 1350 °C.34 This finding is primarily attributed to the adequate mixing that is needed to promote uniformity of the starting materials, which in turn influences the mullitization temperature and improves the properties of the composite aerogels.
Fig. 7 shows the microstructures of the Al2O3–SiO2 composite aerogels with different Al/Si molar ratios, which are heat-treated at 600 °C. The composite aerogels undergo a continuous structural transition from boehmite to γ-Al2O3 as revealed by XRD and TG/DSC analysis. It is found that with the increase of Al/Si molar ratios, the morphologies gradually change from spherical particles to needle-like reticulated networks. This is consistent with the fact that silica particles are usually mono-dispersed and spherical shape,35 and the alumina aerogels always consist of fibrous particles with varying lengths, interconnected to form a web-like microstructure.36,37 The composite aerogels present homogenous porous structures of a typical colloidal gel, which is consisted of SiO2, γ-Al2O3 nanoparticles and nanopores.38 It is found that the nanoparticles adjoin closely to each other and thus possesses the smallest nanopores with diameter at around 20–30 nm for the sample with AS = 2, when compared with the other samples. There are some large pores in the sample with AS = 3, and some agglomeration particles appear in the sample with AS = 4, which is not beneficial for large specific surface areas at elevated temperatures. On the other hand, the sample with AS = 8 exhibits a significant homogeneous pore structure with diameters in the range of approximately 30–40 nm.
 |
| | Fig. 7 SEM images of Al2O3–SiO2 composite aerogel heat-treated at 600 °C with different Al/Si molar ratios. | |
Fig. 8 shows the TEM and HRTEM images of the Al2O3–SiO2 composite aerogel (AS = 8) that is heat-treated at different temperatures. The as-dried Al2O3–SiO2 composite aerogel via supercritical C2H5OH fluid drying shows a sparse structure that consists of needle-like boehmite and amorphous silica. As shown in Fig. 8(b), γ-Al2O3 appears in the Al2O3–SiO2 composite aerogel when it is heat-treated at 600 °C. It possesses microstructures that contain highly reticulated networks of leaflets or sheets, with varying widths of several nanometers and varying lengths, whereas amorphous SiO2 consists of connected spheroidal particles that surround the fibrous γ-Al2O3. With a further heat treatment at 1000 °C, the quantity and size of fibrous γ-Al2O3 increases, indicating the structural transition and normal phenomenon of grain growth occur after treatment at higher temperatures. Electron diffraction spots are not evidently observed in diffraction patterns (shown as inlet in Fig. 8(a)–(c)), which indicates that the composite remains amorphous after heat treatment at maximum temperatures of 1000 °C, which is consistent with the results of XRD patterns. With a higher heat treatment at 1200 °C for 2 h, the composite aerogel is composed of significantly thicker rod-like particles corresponding to γ-Al2O3, and there appears the mullite phase with thicknesses in the range from 20 to 30 nm and lengths that range from of 40 to 80 nm. The diffraction patterns show an intense electron diffraction spot, which also confirms the generation of mullite particles and this is consistent with XRD analysis. Note that the composite heat-treated at 1200 °C is primarily composed of the mullite and γ-Al2O3 phase. This phenomenon verifies that alumina particle growth after heat treatment is restricted after doping with silica. Because γ-Al2O3 usually transforms to δ-Al2O3 or θ-Al2O3 at temperatures greater than 1000 °C.20,39 With an increase in heat treatment temperatures at 1300 °C, there are almost no other forms except the mullite phase and the diameter of the majority of the mullite particles range from 20 to 50 nm. The crystallite size of the mullite phase derived from the heat treatment at 1300 °C is 23.7 nm by analyzing the profile of the diffraction peaks using Scherrer relation equation, which is consistent with the TEM results. Some agglomerates exist in the powders, which is attributed to uncontrolled coagulation during synthesis and sintering. Selected area electron diffraction (SAED) patterns are shown as an inlet in Fig. 8(e), which exhibits a large degree of crystallinity compared with the former samples because the normal phenomenon of mullite grain growth occurs after heat treatment at higher temperatures. In combination with the XRD patterns and the SAED rings, the lattice fringe in Fig. 8(f) also reveals the resulting product is mullite. The lattice fringe is so evident, which is caused by the normal growth of mullite particles, and the distinct lattice fringe with an interplane distance of 0.381 nm can be clearly indexed to the (200) plane of mullite. However, no other forms of lattice fringes have been detected because the mullite phase is the main phase of the mixture after heat treatment at 1300 °C, which obscures the other crystal forms.
 |
| | Fig. 8 TEM images, SAED patterns and HRTEM image of Al2O3–SiO2 composite aerogel (AS = 8) heat-treated at different temperatures: (a) as-dried, (b) 600 °C, (c) 1000 °C, (d) 1200 °C and (e and f) 1300 °C. | |
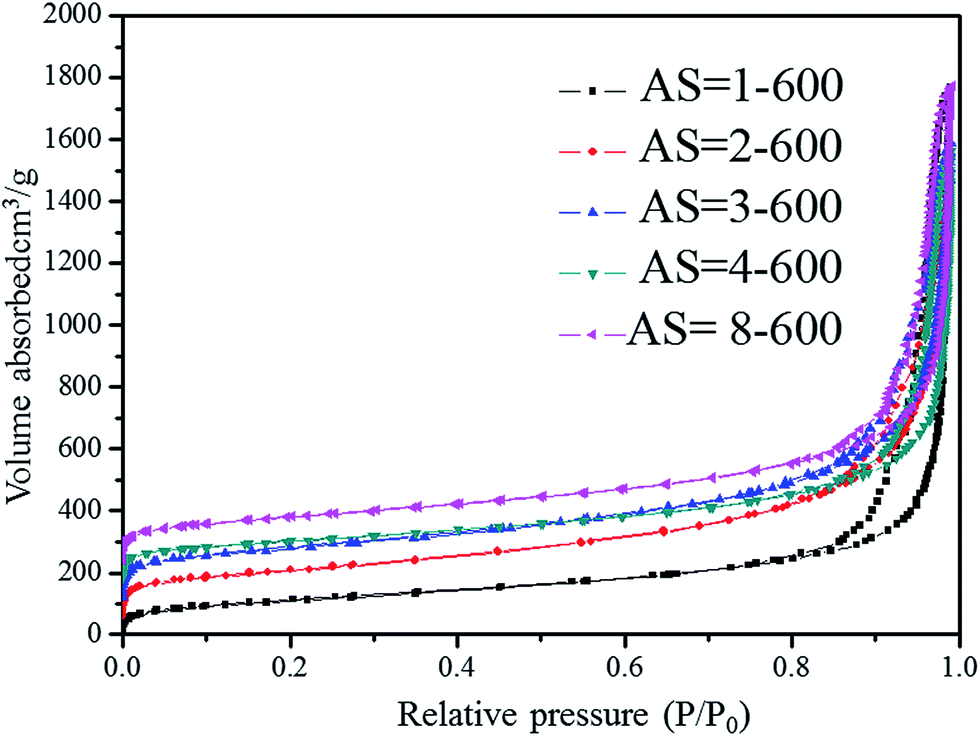
Fig. 9 and 10 show the nitrogen sorption isotherms of Al2O3–SiO2 composite aerogels with different Al/Si molar ratios, which are heat-treated at 600 °C and 1200 °C, respectively. They are type IV curves with type H1 hysteresis loop in the IUPAC classification, which is characteristic of a mesoporous structure with cylindrical pores.38 The desorption cycles of the isotherms show a hysteresis loop for the five samples, which is generally attributed to the capillary condensation that occurs in the mesopores.40 As is well known, the specific surface area is calculated by eqn (7) as follows:
| |
 | (7) |
where
Am is the cross-sectional area of hexagonal close-packed liquid nitrogen (0.162 nm
2),
NA is the Avogadro constant (6.02 × 10
23/mol), and
Vm is the saturated adsorption capacity of the absorbent. Thus, the specific surface area is positively correlated with the saturated adsorption capacity. As is shown in
Tables 1 and
2, the specific surface areas undergo an increase–decrease–increase trend with the increase of Al/Si molar ratios. This is because the fact that with the increase of Al/Si molar ratios, the pore structures of the composite become more homogenous and some small mesoporous with diameters at around 10 nm appear in the composite (as reveals by Fig. S3
†), which is favorable to increasing the specific surface areas of the composites. However, when the Al/Si molar ratio is 4, there appears some agglomeration particles in the composite, which results in the decrease of specific surface area, as indicated by the SEM images in
Fig. 7. The average diameters are below 20 nm except the sample with AS = 1, whereas the composite possesses the minimum average pore diameter of the sample with AS = 2, which is also consistent with the SEM analysis. All the samples heat-treated at 600 °C have pore volumes more than 2 g cm
−3, which are larger than the Al
2O
3–SiO
2 composite aerogel, as reported by Li
41 et al. With a further heat treatment at 1200 °C, the specific surface areas decrease with an increase in heat treatment temperatures, due to the structural adjustment, damage to the porous network and shrinkage of the pore structure. However, they still exhibit the characteristic of an increase–decrease–increase trend with the increase of Al/Si molar ratios. Note that the sample with AS = 3 possesses a significantly larger volume adsorbed (vertical coordinate) than other samples; however, it doesn't mean that the sample possesses the largest specific surface area since the specific surface area is correlated with the saturated adsorption capacity rather than volume adsorbed (as indicated by
eqn (7)). It is found that the sample has the largest specific surface area (166 m
2 g
−1) with AS = 8, compared with other samples. The pore volume of the sample with AS = 3 is as large as 1.4822 g cm
−3, which is much larger than the other samples. This is due to the appearance of some large pores in the composite, because the average pore diameter is 55.681 nm, which exceeds the extent of mesoporous (2–50 nm). The sample with AS = 8 has a rather small average pore diameter (14.380 nm) and possesses a relatively large pore volume (0.5979 g cm
−3). The specific surface area of the sample with AS = 8 is largest among the five samples and it is larger than pure Al
2O
3 aerogel, as reported by Poco
16 (376 m
2 g
−1 for the as-dried, alkoxide method using less than the stoichiometric amount of water instead of a large water excess)
et al. and Li
41 (328 m
2 g
−1 for the as-dried, aluminum
sec-butoxide as the alumina source and tetraethoxysilane as the silicon source)
et al., and the specific surface area of Al
2O
3–SiO
2 composite aerogels, as reported by Osaki
12 (47 m
2 g
−1 at 1200 °C, aluminum isopropoxide as the alumina source and tetraethoxysilane as the silicon source)
et al. and Feng
11 (116 m
2 g
−1 at 1200 °C, aluminum
sec-butoxide as the alumina source and tetraethoxysilane as the silicon source)
et al. As is well known, the chemical potentials of the convex and concave surfaces (
μ1 and
μ2, respectively) could be calculated as follows:
7where
Ω is the atom volume,
γ is the surface free energy,
ρ is the surface curvature and
μ0 is the chemical potential of the flat surface. The chemical potential of the convex surface is larger than the concave's, thus the atom migration occurs from the convex to the concave spontaneously to reach steady state. Alumina particles are connected by “necks”, thus it is favorable for rearrangement of atoms and α-phase nucleation is easy to occur at the neck.
1 According to molecular dynamics, when the cation vacancy of γ-Al
2O
3 is substituted by silicon atom, a protective layer would form in the surface. It is well known that the γ-Al
2O
3 belongs to spinel structure with deficient which possesses eight tetrahedral cation positions, combined with sixteen octahedral cation positions and thirty-two octahedral anion positions per structural unit.
11 Thus, there are as many as eight-thirds vacancies per structural unit within the structure of γ-Al
2O
3, and the vacancy ratio is one-ninth, in comparison with the normal structure. Thus the silicon atoms can easily act as the interstitial atom in the structure. This phenomenon inhibits the diffusivity of alumina atoms and hinders the formation of the contacts and necks between alumna particles, which is favorable to decreasing the probability of crystal nucleation and α-Al
2O
3 phase transition. When the composite aerogels are used as thermal insulations, they are enhanced by the mullite fiber mat to improve the poor mechanical properties. As is shown in Table S2,
† the thermal conductivities of the three samples are 0.023 W m
−1 K
−1, 0.029 W m
−1 K
−1 and 0.025 W m
−1 K
−1, respectively. They are comparable to Al
2O
3 aerogel prepared with aluminum tri-
sec-butoxide reported by Poco
16 et al. (0.029 W m
−1 K
−1 at 30 °C) and Zu
3 et al. (0.028 W m
−1 K
−1 at 30 °C), which appears suitable for efficient thermal insulation uses.
 |
| | Fig. 9 Nitrogen sorption isotherms of Al2O3–SiO2 composite aerogel with different Al/Si molar ratios heat-treated at 600 °C. | |
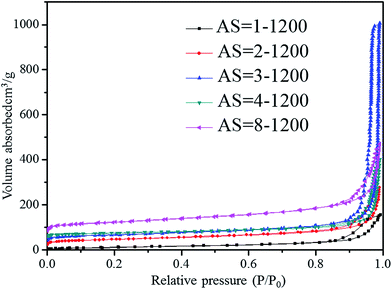 |
| | Fig. 10 Nitrogen sorption isotherms of Al2O3–SiO2 composite aerogel with different Al/Si molar ratios heat-treated at 1200 °C. | |
Table 1 Pore structures of Al2O3–SiO2 composite aerogels with different Al/Si molar ratios heat-treated at 600 °C
| Al/Si molar ratios |
Specific surface area (m2 g−1) |
Average pore diameter (nm) |
Pore volume (cm3 g−1) |
Saturated adsorption capacity (cm3 g−1) |
| 1 |
412.76 |
26.521 |
2.7367 |
94.834 |
| 2 |
536.94 |
15.502 |
2.0943 |
123.36 |
| 3 |
583.09 |
15.562 |
2.2685 |
133.97 |
| 4 |
440.14 |
19.405 |
2.1352 |
101.12 |
| 8 |
511.86 |
18.499 |
2.3672 |
117.60 |
Table 2 Pore structures of Al2O3–SiO2 composite aerogels with different Al/Si molar ratios heat-treated at 1200 °C
| Al/Si molar ratios |
Specific surface area (m2 g−1) |
Average pore diameter (nm) |
Pore volume (cm3 g−1) |
Saturated adsorption capacity (cm3 g−1) |
| 1 |
49.75 |
18.924 |
0.2354 |
11.430 |
| 2 |
98.514 |
16.196 |
0.3989 |
22.634 |
| 3 |
106.48 |
55.681 |
1.4822 |
24.500 |
| 4 |
60.539 |
34.596 |
0.5236 |
13.909 |
| 8 |
166.30 |
14.380 |
0.5979 |
38.208 |
4 Conclusions
A new, inexpensive, and straightforward sol–gel route to synthesise Al2O3–SiO2 composite aerogel with different Al/Si molar ratios using inexpensive inorganic salt of aluminum is investigated. Results show that SiO2 is essentially amorphous, whereas Al2O3 predominately exists as polycrystalline boehmite. With the increase of Al/Si molar ratios, the morphologies change from connected spheroidal particles to nanometer-sized fibrous particles and weblike microstructures. With a higher heat treatment temperature at 600 °C, the structural transition from boehmite to γ-Al2O3 occurs within the composite aerogels, and mullitization firstly occurs with AS = 1 at ca. 1000 °C. The specific surface area undergoes an increase–decrease–increase process at 600 °C and 1200 °C for the composite aerogels with different Al/Si molar ratios. Note that the specific surface area is as high as 166 m2 g−1 at 1200 °C for the sample with AS = 8, which indicates that proper doped silica can effectively inhibit the α-Al2O3 phase transition. The thermal conductivities of the mullite fiber mat reinforced aerogel composites are 0.023 W m−1 K−1, 0.029 W m−1 K−1 and 0.025 W m−1 K−1, suitable for efficient thermal insulations uses.
Acknowledgements
This work was financially supported by the Industry Program of Science and Technology Support Project of Jiangsu Province (BE2014128), the clinical medical special Program of Science and Technology Project of Jiangsu Province (BL2014074), the Prospective Joint Research Program of Jiangsu Province (BY2015005-01), the Major Program of Natural Science Fund in Colleges and Universities of Jiangsu Province (15KJA430005), China Postdoctoral Science Foundation (2015M570442), the Aeronautical Science Foundation of China (201452T4001), the Program for Innovative Research Team in University of Ministry of Education of China (No. IRT_15R35), China Postdoctoral Science Foundation (2015M570442), Jiangsu Collaborative Innovation Center for Advanced Inorganic Function Composites and the Priority Academic Program Development of Jiangsu Higher Education Institutions. Any opinions, findings, and conclusions or recommendations expressed in this paper are those of the authors and do not necessarily reflect the views of these programs.
References
- G. Q. Zu, J. Shen, L. P. Zou, W. Q. Wang, Y. Lian, Z. H. Zhang and A. Du, Chem. Mater., 2013, 25, 4757–4764 CrossRef CAS.
- X. D. Wu, Y. Zhong, Y. Kong, G. F. Shao, S. Cui, L. Wang, J. Jiao and X. D. Shen, J. Porous Mater., 2015, 22, 1235–1243 CrossRef CAS.
- G. Q. Zu, J. Shen, X. Q. Wei, X. Y. Ni, Z. H. Zhang, J. C. Wang and G. W. Liu, J. Non-Cryst. Solids, 2011, 357, 2903–2906 CrossRef CAS.
- E. Ponthieu, J. Grimblot, E. Elaloui and G. M. Pajonk, J. Mater. Chem., 1993, 3, 287–293 RSC.
- N. A. Yassir and R. L. Mao, Appl. Catal., A, 2007, 317, 275–283 CrossRef.
- W. Q. Wang, Z. H. Zhang, G. Q. Zu, J. Shen, L. P. Zou, Y. Lian, B. Liu and F. Zhang, RSC Adv., 2014, 4, 54864–54871 RSC.
- T. Horiuchi, T. Osaki, T. Sugiyama, K. Suzuki and T. Mori, J. Non-Cryst. Solids, 2001, 291, 187–198 CrossRef CAS.
- T. Horiuchi, T. Osaki, T. Sugiyama, H. Masuda, M. Horio, K. Suzuki, T. Mori and T. Sago, J. Chem. Soc., Faraday Trans., 1994, 90, 2573–2578 RSC.
- A. Douy, J. Eur. Ceram. Soc., 2006, 26, 1447–1454 CrossRef CAS.
- L. Xu, Y. G. Jiang, J. Z. Feng, J. Feng and C. W. Yue, Ceram. Int., 2015, 41, 437–442 CrossRef CAS.
- J. Feng, Q. F. Gao, W. Wu, C. R. Zhang, J. Z. Feng and Y. G. Jiang, Chin. J. Inorg. Chem., 2009, 25, 1758–1763 CAS.
- T. Osaki, K. Nagashima, K. Watari and K. Tajiri, J. Non-Cryst. Solids, 2007, 353, 2436–2442 CrossRef CAS.
- B. E. Yoldas, J. Mater. Sci., 1975, 10, 1856–1860 CrossRef CAS.
- M. S. Bono, A. M. Anderson and M. K. Carroll, J. Sol-Gel Sci. Technol., 2010, 53, 216–226 CrossRef CAS.
- L. N. Wu, Y. D. Huang, Z. J. Wang, L. Liu and H. F Xu, Appl. Surf. Sci., 2010, 256, 5973–5977 CrossRef CAS.
- J. F. Poco, J. H. Satcher and L. W. Hrubesh, J. Non-Cryst. Solids, 2001, 285, 57–63 CrossRef CAS.
- F. I. Hurwitz, M. Gallagher, T. C. Olin, M. K. Shave, M. A. Ittes, K. N. Olafson, M. G. Fields and R. B. Rogers, Int. J. Appl. Glass Sci., 2014, 5, 276–286 CrossRef CAS.
- G. Q. Zu, J. Shen, W. Q. Wang, L. P. Zou, Y. Lian and Z. H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 5400–5409 CAS.
- A. E. Gash, J. H. Satcher and R. L. Simpson, Chem. Mater., 2003, 15, 3268–3275 CrossRef CAS.
- F. C. Cao, L. L. Ren and X. A. Li, RSC Adv., 2015, 5, 18025–18028 RSC.
- A. Du, B. Zhou, Z. H. Zhang and J. Shen, Materials, 2013, 6, 941–968 CrossRef CAS.
- G. Q. Zu, J. Shen, W. Q. Wang, L. P. Zou, Y. Lian, Z. H. Zhang, B. Liu and F. Zhang, Chem. Mater., 2014, 26, 5761–5772 CrossRef CAS.
- G. Q. Zu, J. Shen, W. Q. Wang, L. P. Zou, W. W. Xu and Z. H. Zhang, Acta Phys.-Chim. Sin., 2015, 31, 360–368 CAS.
- K. Okada, C. Aoki, T. Ban, S. Hayashi and A. Yasumori, J. Eur. Ceram. Soc., 1996, 16, 149–153 CrossRef CAS.
- S. Komarneni and C. Rutiser, J. Eur. Ceram. Soc., 1996, 16, 143–147 CrossRef CAS.
- L. Xu, Y. G. Jiang, J. Z. Feng and J. Feng, Mater. Sci. Forum, 2015, 816, 157–162 CrossRef.
- W. A. Yarbrough and R. Roy, J. Mater. Res., 1987, 2, 494–515 CrossRef CAS.
- H. W. Hou, Y. Xie, Q. Yang, Q. X. Guo and C. R. Tan, Nanotechnology, 2005, 16, 741–745 CrossRef CAS.
- Q. P. Wang, X. L. Li, W. P. Fen, H. M. Ji, X. H. Sun and R. Xiong, J. Porous Mater., 2014, 21, 127–130 CrossRef CAS.
- D. Sarkar, D. Mohapatra, S. Ray, S. Bhattacharyya, S. Adak and N. Mitra, Ceram. Int., 2007, 33, 1275–1282 CrossRef CAS.
- D. Y. Li, Y. S. Lin, Y. C. Li, D. L. Shieh and J. L. Lin, Microporous Mesoporous Mater., 2008, 108, 276–282 CrossRef CAS.
- W. M. Kriven, L. F. Siah, M. Schmücker and H. Schneider, J. Am. Ceram. Soc., 2004, 87, 970–972 CrossRef CAS.
- A. P. Hynes and R. H. Doremus, J. Am. Ceram. Soc., 1991, 74, 2469–2475 CrossRef CAS.
- J. C. Huling and G. L. Messing, J. Am. Ceram. Soc., 1991, 74, 2374–2381 CrossRef CAS.
- Y. Fazli, E. Kulani, K. Khezri and H. Alijani, Microporous Mesoporous Mater., 2015, 214, 70–79 CrossRef CAS.
- T. F. Baumann, A. E. Gash, S. C. Chinn and A. M. Sawvel, Chem. Mater., 2005, 17, 395–401 CrossRef CAS.
- C. L. Carnes, P. N. Kapoor, K. J. Klabunde and J. Bonevich, Chem. Mater., 2002, 14, 2922–2929 CrossRef CAS.
- X. B. Yi, L. L. Zhang, F. Y. Wang, X. D. Shen, S. Cui, J. Zhang and X. C. Wang, RSC Adv., 2014, 4, 48601–48605 RSC.
- A. Sedaghat and E. Taheri, J. Adv. Mater. Process., 2014, 2, 25–32 Search PubMed.
- J. S. Lee and S. C. Yu, J. Mater. Sci., 1992, 27, 5203–5208 CrossRef CAS.
- X. Li, G. T. Qin, Y. T. Wang and W. Wei, J. Sol-Gel Sci. Technol., 2014, 72, 405–414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19764c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.