
Synthesis and electrochemical properties of monoclinic fluorine-doped lithium manganese oxide (LixMnO2−yFy) for lithium secondary batteries
Yu Zhang,
Zhi Su*,
Xiang Yao and
YingBo Wang
College of Chemistry and Chemical Engineering, Xinjiang Normal University, Urumqi, 830054, Xinjiang, China. E-mail: suzhixj@sina.com; Fax: +86-991-4332683; Tel: +86-991-4332683
First published on 14th October 2015
Abstract
A series of monoclinic fluorine-doped lithium manganese oxide (LixMnO2−yFy) were prepared by the ion exchange of sodium for lithium in NaxMnO2−yFy precursors that were obtained using a high-temperature solid-state reaction. The microstructure and composition of the samples were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), inductively coupled plasma atomic emission spectroscopy (ICP-AES), ion chromatography (IC), Fourier transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS). The different valence states of manganese in material LixMnO2−yFy were determined by redox titration method. The electrochemical performance of these samples as cathode materials were studied by the galvanostatic and cyclic voltammetry method. These materials had a high crystallite size, which was composed of 5–8 μm grains. The Li0.86MnO1.98F0.02 materials delivered an initial discharge capacity of 129.2 mA h g−1 and gradually increased to a maximum discharge capacity of 210 mA h g−1 at a current density of 50 mA g−1 after 50 cycles. Moreover, the material showed an excellent cycling behavior, even though its original structure transformed into the spinel phase during cycling. The results show that the partial substitution of monoclinic LiMnO2 with fluorine can improve the cycle stability and high-rate discharge capability of cathode materials.
1. Introduction
Lithium manganese oxide (LiMnO2) compounds have received attention as intercalation cathodes for rechargeable lithium ion batteries due to their high theoretical capacity, low cost, and nontoxicity, and there has been significant interest in monoclinic LiMnO2 (space group C2/m, hereafter denoted as m-LiMnO2) as a potential cathode material for replacing LiCoO2, which is the currently used cathode material in commercial Li-ion rechargeable batteries.1–3 However, m-LiMnO2 is generally considered to be unstable compared to orthorhombic LiMnO2 (space group Pmnm, hereafter denoted as o-LiMnO2).4 One significant drawback is the difficult synthesis of layered m-LiMnO2 using the conventional high-temperature solid-state reaction method because these methods always produce o-LiMnO2.5,6 Consequently, m-LiMnO2 was first obtained by metastable synthesis routes such as ion exchange and hydrothermal reactions.7,8In this compound, the O2− ions are arranged in a cubic close-packed structure, and the octahedral interstices are occupied by Li+ and Mn3+ with layered cation ordering. The neighboring MnO6 octahedra share a common edge. Due to the presence of high-spin Mn3+ on the octahedral sites, the local site symmetry around Mn3+ is distorted from a regular octahedron, and the overall crystal structure is monoclinic due to the cooperative Jahn–Teller distortion. Therefore, it is difficult to inhibit the irreversible structural transformation from layered LiMnO2 to a spinel LiMnO2 during electrochemical cycling. According to the first-principles calculations, the Jahn–Teller effect should be absent in the low-spin state, and the layered structure could be stabilized in m-LiMnO2.9
Cation doping protects the layered monoclinic structure against Jahn–Teller distortion10 and has superior Li insertion/extraction cycling properties for m-LiMnO2. Many studies have focused on stabilization of the layered material structure, particularly for m-LiMnO2, by partial substitution of Mn by Cr, Al, Co, Ni and Mg.11–18 However, for m-LiMnO2 to be suitable for practical applications such as high-energy and power density applications, further stabilization of the monoclinic structure is needed. Small amounts of additional dopants such as fluorine19 into the Li(Ni1/3Co1/3Mn1/3)O2 lattice have been reported to improve the structural stability and enhance the cycling performance. Zheng et al. prepared the Li[Li0.2Mn0.54Ni0.13Co0.13]O2−xFx (x = 0, 0.05 and 0.10) composite materials with different content of fluorine by sol–gel method using NH4F as fluorine source. XRD and XPS analysis shows that the fluorine ions successfully substitute for oxygen sites. The F-doped Li[Li0.2Mn0.54Ni0.13Co0.13]O2−xFx materials show superior cycling performance.20 In fact, Jouanneau et al. discussed the influence of LiF additions on Li(NixCo1−2xMnx)O2 and found that materials could be obtained using LiF as a sintering agent with almost no disturbance of the cell performance.21–23
In this work, we attempt to prepare a partial substitution of O2− by F− layered monoclinic LiMnO2 using a high-temperature solid-state reaction method with a subsequent ion-exchange reaction. The morphology, microstructure, composition, and electrochemical properties were investigated in detail to assess the suitability of these samples as cathode materials.
2. Experimental
2.1 Preparation and characterization
First, Mn(CH3COO)2·4H2O was heated with the ramping rate of 2 °C min−1 and decomposed at 500 °C for 15 h in air to produce Mn2O3. Samples of monoclinic α-NaMnO2 were prepared using a solid-state reaction of the stoichiometric mixture of Mn2O3 (0.01 mol, 1.58 g) and Na2CO3 (Na/Mn = 1.1, 0.011 mol, 1.16 g) in a closed crucible at 750 °C for 24 h in air. The mixture was quenched to room temperature and ground, and the process was repeated. The resulting samples were stored in a desiccator. In the subsequent ion-exchange reaction, 1 g of α-NaMnO2 was mixed with 10 g LiCl in 40 ml n-hexanol and autoclaved at 150 °C for 12 h in a 50 ml Teflon-lined stainless steel vessel. The obtained m-LiMnO2 products were washed with de-ionized water and then dried in an oven at 105 °C for 12 h. Fluorine-substituted materials were prepared with the molar ratio of F/Mn of 0.01/1, 0.03/1, and 0.05/1 with the homogeneous mixture of NH4F of 0.001 mol (0.037 g); 0.003 mol (0.0111 g); and 0.005 mol (0.0185 g), respectively. The final fluorine-substituted materials were obtained by the above-mentioned method.The microstructure of the as-prepared samples was characterized by X-ray diffraction (XRD, Rigaku D/max-2500) with CuKα radiation and scanning electron microscopy (SEM, HITACHI S-3500N). Elemental analysis for Na, Li, and Mn were performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES, IRIS Advantage) and the content of fluorine by ion chromatography (ICS-2100) to determine the chemical composition of these materials. The Fourier transform infrared spectra (FTIR) of these samples were recorded from 400 to 4000 cm−1 at room temperature using the KBr wafer technique in a Bio-Rad FTS 6000 FTIR instrument with a resolution of 2 cm−1 in transmittance mode. The surface chemical state was recorded using X-ray photoelectron spectroscopy (XPS, PHI-5300 ESCA).
2.2 Determination of material composition
The sample was treated by HCl, HNO3 and HF at room temperature, H3BO3 was added to complex with excessive fluoride ions. Working conditions of the instrument was optimized, appropriate analysis line of various elements was selected and the internal standard method was used to eliminate disturbance with yttrium selected as internal standard element, the content of lithium and manganese were determined by inductively coupled plasma emission spectroscopy (ICP-AES). The sample was decomposed by H2SO4. After water steam distillation at 160–180 °C, the fluoride in testing solution would be released and absorbed by NaOH solution realizing the separation from matrix and other elements. Then, the content of fluoride was determined by ion chromatography (IC).2.3 Analysis of the valence states of manganese
The total amount of manganese was determined by chemical conversion to permanganic radical using acidified ammonium persulfate, the determination of Mn4+ by the acetylacetone-iodometric method being carried out in the presence of Mn3+ without its interference, and the amount of the tetravalent and trivalent manganese being determined by dissolving the lithium manganese oxides in a known excess of ammonium iron sulfate in 1 mol L−1 sulfuric acid and back-titrating the unreacted Fe2+ with standardized potassium dichromate. Based on the results obtained, the amount of bivalent manganese and average manganic valence could be calculated.242.4 Electrochemical performance
The working electrode was prepared by compressing a mixture of the active materials with acetylene black and a binder (polytetrafluoroethylene, PTFE) in a weight ratio of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:5. Lithium metal was used for the counter and reference electrodes. The electrolyte was LiPF6 (1 M) in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) in a weight ratio of 1:1:1. All procedures for handing and fabricating the electrochemical cells were performed in an argon-filled glove box. The galvanostatic method was used to measure the electrochemical capacity and cycle life of the electrodes under the charge–discharge current density of 50 mA g−1 at room temperature using a LAND CT2001A instrument. The cut-off potentials for the charge and discharge were set between 2.0–4.3 V (vs. Li+/Li). Cyclic voltammetry (CV) was performed with a LK2500 electrochemical workstation, scanning from 2 to 4.3 V with a scan rate of 0.1 mV s−1. The electrochemical impedance spectra (EIS) were measured using a Zahner IM6ex electrochemical workstation over the frequency ranges from 10 kHz to 10 MHz with a 5 mV AC input signal applied between the working and reference electrodes.
:20:5. Lithium metal was used for the counter and reference electrodes. The electrolyte was LiPF6 (1 M) in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) in a weight ratio of 1:1:1. All procedures for handing and fabricating the electrochemical cells were performed in an argon-filled glove box. The galvanostatic method was used to measure the electrochemical capacity and cycle life of the electrodes under the charge–discharge current density of 50 mA g−1 at room temperature using a LAND CT2001A instrument. The cut-off potentials for the charge and discharge were set between 2.0–4.3 V (vs. Li+/Li). Cyclic voltammetry (CV) was performed with a LK2500 electrochemical workstation, scanning from 2 to 4.3 V with a scan rate of 0.1 mV s−1. The electrochemical impedance spectra (EIS) were measured using a Zahner IM6ex electrochemical workstation over the frequency ranges from 10 kHz to 10 MHz with a 5 mV AC input signal applied between the working and reference electrodes.
3. Results and discussion
3.1 Composition and structure
The composition of the as-prepared samples, which were calculated from the results of ICP-AES, and IC determination and valence state of Mn analysis, are summarized in Table 1. For all samples, most of the Na+ cation can be exchanged and lithium concentration can be controlled at about 0.90 in unit. All of the XRD patterns were refined by the Rietveld analysis, an example of the Rietveld refinement is shown in Fig. 1 for the composition of LixMnO2−yFy. All diffraction peaks, except for the market peaks, could be attributed to the small amount of monoclinic Na0.7MnO2.05 because NaMnO2 was not stable and was very hygroscopic in air; NaMnO2 was easily transformed to the more stable phase of Na0.7MnO2.05.25| Sample | Design formula | Precursor powders | Valence of manganese | Chemical composition |
|---|---|---|---|---|
| a | NaMnO2 | Na0.96MnO2 | 3.04 | Na0.96MnO2 |
| b | LiMnO2 | Na0.91MnO2 | 3.09 | Na0.06Li0.85MnO2 |
| c | LiMnO1.99F0.01 | Na0.90MnO1.98F0.02 | 3.08 | Na0.05Li0.86MnO1.98F0.02 |
| d | LiMnO1.97F0.03 | Na0.91MnO1.96F0.04 | 3.05 | Na0.06Li0.88MnO1.96F0.04 |
| e | LiMnO1.95F0.05 | Na0.92MnO1.93F0.07 | 3.01 | Na0.06Li0.87MnO1.93F0.07 |
 | ||
| Fig. 1 The XRD patterns of the as-prepared samples: (a) Na0.96MnO2, (b) Na0.06Li0.85MnO2, (c) Na0.05Li0.86MnO1.98F0.02, (d) Na0.06Li0.88MnO1.96F0.04, and (e) Na0.06Li0.87MnO1.93F0.07. | ||
The Table 2 list the comparison between the observed values and the JCPDS-87155 standard card data. The characteristics of the synthetic product crystal plane diffraction spacing and diffraction angle all can correspond well, the as-prepared samples could be assigned to the monoclinic structure with space group of C2/m, which differs from o-LiMnO2.7–9 At the same time the characteristic peak of α-NaMnO2 are not appear noticeably in XRD, this shows that ion exchange basic completely. Moreover, we also find that the observed dhkl values is greater than the standard and the 2θ values is opposite. This may be due to the material contains no exchange of sodium (sodium ions with greater radius than lithium ion).
| h k l | dhkl (LiMnO2)/nm | 2θ/(°) | ||
|---|---|---|---|---|
| Observed | JCPDS | Observed | JCPDS | |
| 0 0 1 | 0.4849 | 0.4842 | 18.28 | 18.31 |
| −2 0 1 | 0.2712 | 0.2711 | 33.02 | 33.01 |
| 1 1 0 | 0.2440 | 0.2435 | 36.80 | 36.89 |
| −1 1 1 | 0.2397 | 0.2395 | 37.48 | 37.50 |
| −2 0 2 | 0.2298 | 0.2295 | 39.12 | 39.21 |
| 1 1 1 | 0.2012 | 0.2006 | 45.02 | 45.15 |
| −1 1 2 | 0.1942 | 0.1942 | 46.46 | 46.74 |
| 2 0 1 | 0.1878 | 0.1875 | 48.45 | 48.49 |
| 0 0 3 | 0.1626 | 0.1614 | 56.52 | 57.01 |
| 1 1 2 | 0.1560 | 0.1555 | 59.16 | 59.37 |
| −1 1 3 | 0.1511 | 0.1505 | 61.26 | 61.56 |
| −3 1 2 | 0.1493 | 0.1487 | 62.12 | 62.36 |
| 0 2 0 | 0.1407 | 0.1404 | 66.34 | 66.53 |
| 0 2 1 | 0.1350 | 0.1348 | 69.54 | 69.66 |
Lattice parameters of all samples obtained from the Rietveld refinement are listed in Table 3. With increasing fluorine content, cell volume increased from 0.0824 nm3 to 0.0839 nm3, the increase of cell volume is attributed to the replacement of O2− (r = 0.132 nm) with greater F− (r = 0.136 nm), which are in agreement with literature values.26 From the table can be found that the increase of the content of fluorine can decrease the valence state of Mn in material. The results attributable to the low valence state of anion (F−) supersedes the high valence state O2−, and it also demonstrated that the fluorine ions successfully doped into the lattice structure.
| Sample | a (nm) | b (nm) | c (nm) | Cell volume (nm3) |
|---|---|---|---|---|
| Na0.96MnO2 | 0.582 | 0.287 | 0.576 | 0.0962 |
| Na0.06Li0.85MnO2 | 0.544 | 0.281 | 0.539 | 0.0824 |
| Na0.05Li0.86MnO1.98F0.02 | 0.543 | 0.282 | 0.541 | 0.0828 |
| Na0.06Li0.88MnO1.96F0.04 | 0.543 | 0.282 | 0.543 | 0.0831 |
| Na0.06Li0.87MnO1.93F0.07 | 0.544 | 0.283 | 0.549 | 0.0839 |
The FTIR spectra of these as-prepared samples are indicated in Fig. 2. The bands in the region of 3000–3500 cm−1 can be assigned to OH stretching vibrations. Moreover, the bending mode vibration of water is observed at 1600–1800 cm−1. The weak absorptions at 2832 and 2886 cm−1 are due to C–H stretching models, and the bands at around 1475 and 2465 cm−1 result from the absorption of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.27 Two obvious absorption bands are observed in the infrared (IR) spectra of these materials at around 875 and 993 cm−1 (except for Na0.96MnO2 samples), which could be attributed to the metal hydroxide vibration band.28 Furthermore, the IR bands in the region of 450–750 cm−1 could be assigned to Mn–O stretching vibrations in lithium-manganese-oxide compounds.29
O group.27 Two obvious absorption bands are observed in the infrared (IR) spectra of these materials at around 875 and 993 cm−1 (except for Na0.96MnO2 samples), which could be attributed to the metal hydroxide vibration band.28 Furthermore, the IR bands in the region of 450–750 cm−1 could be assigned to Mn–O stretching vibrations in lithium-manganese-oxide compounds.29
 | ||
| Fig. 2 The IR spectra of (a) Na0.97MnO2, (b) Na0.07Li0.85MnO2, and (c) Na0.06Li0.86MnO1.98F0.02. | ||
For the Na0.96MnO2 and Na0.06Li0.85MnO2 samples, one featured band at 530 cm−1 is observed. However, for the F-doped sample of Na0.05Li0.86MnO1.98F0.02, an obvious blue shift is observed in the Mn–O stretching band from 532 to 550 cm−1. The electronegativity of F (4.0), which is higher than that of O (3.5), can strengthen the Mn–O stretching bands, whereas the thermochemical radius of F− (0.136 nm) is greater than the O2− ionic radius (0.132 nm), leading to the blue shift.30 Certainly, with the fluoride concentration increase, the blue shift will become more obviously which causes the oxygen defects concentration increase in the material. Due to the deficiency of oxygen ion in the electrode material caused by oxygen defects, there are positive charged oxygen vacancies in the structure; and then during the immersion of electrolytic solution to electrode surface, more oxygen defects produce thicker organic electrolytic liquid film, which arise the organic electrolytic liquid film resistance. This result implies that oxygen defects will reduce the electrochemical capacity and cycling performance of the electrode material; and this is in agreement with the electrochemical performance test results shown below.31
Fig. 3 shows the SEM images of the as-prepared samples. The layered Na0.97MnO2 and monoclinic LixMnO2−yFy samples have a similar bar-shaped morphology with a grain diameter of 5–8 μm and with small particles of about 2–3 μm in size. However, a topotactic reaction can be observed in a morphology changes from the SEM. Small particles can be found in Fig. 3. The things is especially obvious with the increase of fluorine content. The results should be attributed to the primary particles become well-developed when the amount of fluorine increases. Changes of morphology reveal that incorporation of fluorine alters the surface energy and catalyzes the growth of the primary particles, consistent with that reported for other cathode materials with fluorine doping.32,33
 | ||
| Fig. 3 The SEM images of the as-prepared samples: (a) Na0.96MnO2, (b) Na0.06Li0.85MnO2, (c) Na0.05Li0.86MnO1.98F0.02, (d) Na0.06Li0.88MnO1.96F0.04, and (e) Na0.06Li0.87MnO1.93F0.07. | ||
3.2 Electrochemical properties
Fig. 4 exhibits the initial charge/discharge curves of LixMnO2−yFy (y = 0, 0.02, 0.04, 0.07) at a current density of 50 mA g−1 (0.2 C) between 2.0 and 4.3 V. An initial discharge capacity of 133.2 mA h g−1 is obtained for the Na0.06Li0.85MnO2 sample, which is obviously higher than that of o-LiMnO2 obtained using the conventional high-temperature solid reaction. With the increase of fluorine content, the discharge capacities of the initial discharge capacity is decreased to 129.2 mA h g−1 at 0.2C for Na0.05Li0.86MnO1.98F0.02, the initial discharge capacities of Na0.06Li0.88MnO1.96F0.04 and Na0.06Li0.87MnO1.93F0.07 are 120.6 and 112.3 mA h g−1, respectively. This is due to the existence of the inactive substances fluorine and the atomic weight of fluorine (19 g mol−1) slightly larger than oxygen atomic weight (16 g mol−1). Moreover, fluorine substituted materials exhibit higher charging voltage over the potential range and the discharge plateau is lower than the pristine one, which is an indication of the higher polarization effect. This similar phenomena, which is ascribed to the stronger bonding strength of Li–F (577 kJ mol−1) bond than the Li–O bond (341 kJ mol−1), are also observed and explained by previous researchers.34,35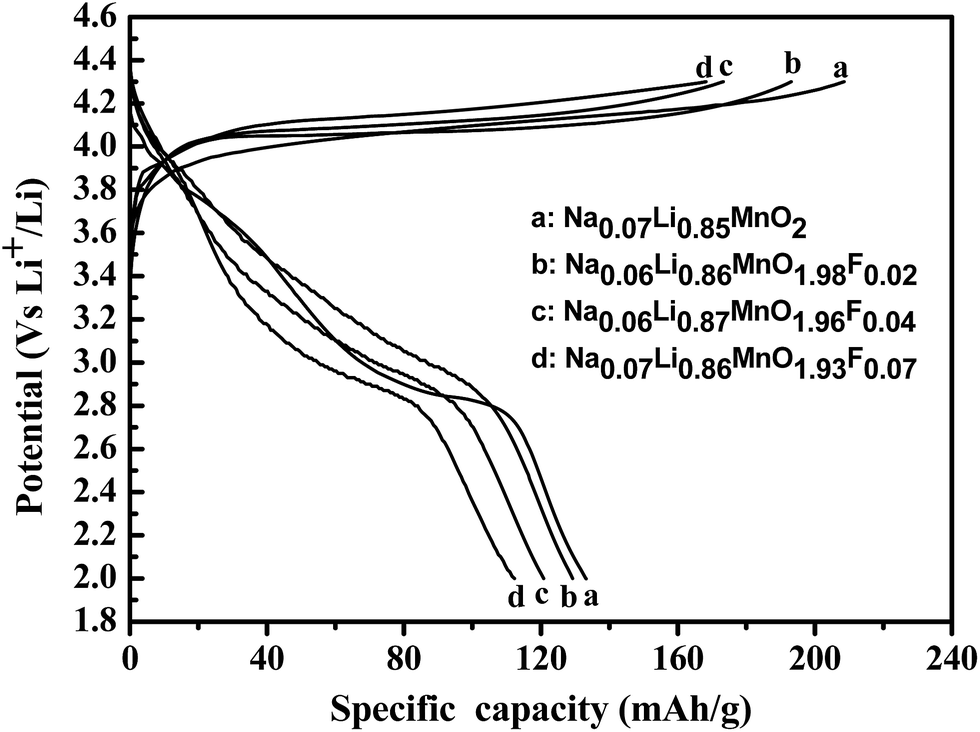 | ||
| Fig. 4 The initial charge–discharge voltage profile (a), differential capacity plots (b) of LixMnO2−yFy (y = 0, 0.02, 0.04, 0.07) when cycled between 2.0 and 4.3 V at a current density of 50 mA g−1. | ||
The rate capabilities of the materials are operated at the same current densities between 2.0 and 4.3 V. Fig. 5 compares the discharge capacities of the electrodes at various C rates. The results of the electrochemical cycles indicated that the discharge capacity of monoclinic LiMnO2 was small at the initial cycles, gradually increased with increasing cycle number, and the capacity finally stabilized. This result was consistent with the previously reported results of orthorhombic LiMnO2 electrochemical cycle behavior. This phenomenon reveals that monoclinic LiMnO2 cathode materials undergo an activation process during the electrochemical cycle, during which there is a phase transition from m-LiMnO2 to cubic spinel LixMn2O4.36 In comparison, the cycle stability of the Na0.06Li0.85MnO2 sample is not so good with a low capacity retention of 80.8 mA h g−1 after 50 cycles. However, the fluorine substituted materials show better rate capability at high charge and discharge rates. The capacity retention of these materials is significantly improved by fluorine substitution, the good rate capability of fluorine substituted materials could be attributed to the existence of the strong bonding by fluorine which stabilizes the host structure and increases the stability of cathode materials.37 The results also show that the electrochemical performances of the materials were severely damaged with the fluorine content increased, this is related to the amount of oxygen defects in materials. Therefore, the fluorine content should be controlled in order to prevent more oxygen defects in synthetic materials. In other words, the partial fluorine substitution in the layered monoclinic structure can significantly depress this structural transformation and slow the rate of capacity decay.
 | ||
| Fig. 5 Cycle performance of the as-prepared samples at different discharge current densities (charging at 50 mA g−1). | ||
We also notice that the Na+ cation can be reserved at about 0.06 unit in all samples. The Na+ ionic radius (0.097 nm) is larger than the Li+ ionic radius (0.060 nm). Thus, in monoclinic Na-substituted LixMnO2, the small change of the interlayer spacing to accommodate Na allows high rechargeability and good cycle life. This result could be explained by the extraction of a limited amount of sodium ions from the interlayer spacing while maintaining the layered structure upon cycling.38
Initial investigation of LixMnO2−yFy were carried out using cyclic voltammetry, Fig. 6. A rate of 0.01 mV s−1 between potential limits of 2 and 4.3 V was employed. Cycling commenced with oxidation from the open circuit potential of 2 V. Fig. 6(a) and (b) shows typical cyclic voltammograms of the Na0.06Li0.85MnO2 and Na0.05Li0.86MnO1.98F0.02 samples subjected to 1, 2, 5, 10, 20 and 30 cycles and 0.1 mV s−1 over the potential range between 2.0 and 4.3 V (vs. Li+/Li). The striking feature is the difference between the first and subsequent cycles. In fact the difference is to be found in the original CV for Na0.06Li0.85MnO2 shows two anodic peaks at 3.25 V and 4.18 V as well as the corresponding cathodic peaks at 2.83 V and 3.85 V. On repeated cycling, the peaks at 4.18 V and 3.85 V split into two peaks, which becomes more obvious after 30 cycles. This peak splitting suggests a gradual transformation of layered LiMnO2 into the spinel phase.34 From Fig. 6(a), we can observe the anodic phase shows four oxidation peaks centered at 3.1 V, 4.0 V, 4.18 V, and 3.8 V after 2 cycles, but the cathodic phase shows only three reduction peaks with a major peak centered at 2.8 V and two minor peaks at 3.9 V and 4.1 V. The peak at 3.8 V does not have reduced peak, which corresponds to an irreversible phase change. Similar irreversible phase changes were observed by Paulsen et al. in O2-type Li2/3[Li1/6Mn5/6]O2 and Li2/3[Co1/18Mn17/18]O2.39 The reason for this irreversible peak/plateau is not presently known; however, because it is only observed in stacking faulted compounds, the irreversible peak could be correlated to crystallographic modification, oxygen removal from the lattice and/or removal of the stacking faults. For the Na0.05Li0.86MnO1.98F0.02 sample, the similar main cathodic and anodic peaks with Na0.06Li0.85MnO2 sample in the first cycle are found in Fig. 6(b). From the CV curves of Fig. 6(b), Li+ can mostly deintercalate from the structure of sample Na0.05Li0.86MnO1.98F0.02 in the first anode process, due to the broader oxidizing peak about 3.8 V. But the reducing peak about 2.9 V is weaker in the first cathode process. This result indicates there is obvious capacity loss between the initial charge and discharge capacities, and the efficiency of reversible intercalate/deintercalate of Li+ is relatively lower, which is in accordance with Fig. 4. As the cycle goes by, the intensity of oxidizing peak about 3 V gradually becomes stronger, and the intensities of reducing peak about 2.8 V and 4 V gradually become stronger. However, the anodic or cathodic current densities change slightly in the following cycles, evidencing the higher electrochemical stability of the Na0.05Li0.86MnO1.98F0.02 sample as compared to the Na0.06Li0.85MnO2 sample.
 | ||
| Fig. 6 Cyclic voltammograms of the (a) Na0.067Li0.85MnO2 and (b) Na0.05Li0.86MnO1.98F0.02 subjected to 1, 2, 5, 10, 20 and 30 cycle. | ||
The Fig. 5 also shows that the variation characteristics of capacity with cycle number for the Na0.06Li0.85MnO2 and Na0.05Li0.86MnO1.98F0.02 materials. The first discharge of the Na0.06Li0.85MnO2 sample is associated with a specific capacity of 133.2 mA h g−1 but this falls significantly to 117.7 mA h g−1 at the third cycle, then slowly increase the 190 mA h g−1 after 20 cycles and the subsequent decay slowly. For the Na0.05Li0.86MnO1.98F0.02 sample, the varying pattern is similar with the Na0.06Li0.85MnO2 sample. The first specific capacity on discharge is 129.2 mA h g−1 but this falls significantly to 95 mA h g−1 at the 5th cycle, and subsequent slowly increase to 219 mA h g−1 after 40 cycles. However, the greatest difference can be found in the thereafter cycle process, the capacity on discharge is very similar at around 218 mA h g−1. This is in accord with the CV in Fig. 6 and reinforces the view that the electrochemical evidence points to a change occurring during the first charge.
In order to further verify the results of electrochemical test and the influence of fluorine doping for the materials. The XRD was used to confirmed by crystal structure measurements of the cycled cathode powder. Fig. 7 shows the XRD pattern of the Na0.067Li0.85MnO2 and Na0.05Li0.86MnO1.98F0.02 subjected to 30 cycles. The diffraction peaks in the XRD pattern broadened after electrochemical cycling, which is attributed to the creation of local lattice strain and tiny crystallites during cycling.40 The results also show that the samples have been converted to the cubic spinel phase LiMn2O4 upon cycling, the results are consistent with the electrochemical experiments. However, the layered structure LiMnO2 phase can obvious exist for the Na0.05Li0.86MnO1.98F0.02 materials in the electrochemical process. This fully shows that fluorine doping can effectively inhibit the structure of the shift.
 | ||
| Fig. 7 XRD patterns of (a) Na0.06Li0.85MnO2 and (b) Na0.05Li0.86MnO1.98F0.02 after 30 cycles. | ||
The Nyquist plots of the electrochemical impedance spectra (EIS) analyses are shown in Fig. 8. All plots consist of both a semicircle in the high-frequency region and a slope in the low-frequency region. The electrode reaction process is primarily controlled by a mixed process of lithium ion diffusion (Warburg impedance, which is inversely proportional to the diffusion coefficient DLi+) and the surface electrochemical reaction at a temporary steady state of EIS. Compared to the Na0.07Li0.85MnO2 sample from Table 4, the Warburg impedance of fluorine-substituted materials is lower, which contributes to their improved high rate capability. On the contrary, the charge-transfer resistance increases after fluorine substitution due to the poor electrochemical activity of the anions, while the Warburg impedance obviously decreases as compared to that of the Na0.07Li0.85MnO2 sample. Clearly, the Warburg impedance of the fluorine-substituted material Na0.06Li0.86MnO1.98F0.02 is much smaller than that of the Na0.07Li0.85MnO2 sample, which is consistent with the improved high-rate capability demonstrated above.
 | ||
| Fig. 8 Electrochemical impedance spectra (EIS) of the as-prepared samples at the discharged state (about 3.0 V (vs. Li+/Li)) after the 30th cycle. | ||
| Sample (ICP-AES) | Rct (Ω) | CPE (×10−5) | Ws (Ω) |
|---|---|---|---|
| a Rct represents the charge-transfer resistance, CPE represents the constant phase element, and Ws represents the Warburg impedance. | |||
| Na0.06Li0.85MnO2 | 122.31 | 1.04 | 270.71 |
| Na0.05Li0.86MnO1.98F0.02 | 132.81 | 1.08 | 36.25 |
| Na0.06Li0.88MnO1.96F0.04 | 152.91 | 1.05 | 112.32 |
| Na0.06Li0.87MnO1.93F0.07 | 221.62 | 2.91 | 117.31 |
3.3 XPS spectra analysis of materials
The XPS spectra can provide chemical information such as the oxidation state as well as the semi-quantitative composition of the surface, thus enabling observation of the surface properties. Fig. 9(a) shows the Mn 2p XPS spectra of the Na0.05Li0.86MnO1.98F0.02 sample. The binding energies of Mn 2P1/2 and Mn 2p3/2 were determined to be 653.86 eV and 641.80 eV, respectively. The peaks at the binding energy of about 641.60 eV are similar to those observed in Mn2O3 and MnOOH.41 The binding energy peak situated at 653.86 is close to the binding energy of Mn 2P1/2 (653.90 eV) observed in the Mn2O3 sample. The binding energy of Na 1s was determined to be 1071.26, which is similar to that observed in NaF.42 Moreover, the binding energy of F 1s is situated at 686.1 eV, which is similar to that observed at 686.1 eV in MnF2.43 To further determine the existence of fluorine within the crystal structure, XPS measurement was carried out on the four samples. Fig. 9(b) shows the XPS spectra of F 1s for the pristine and F-doped materials. All curve fittings were based on C 1s (C–C bonding energy) calibration to 284.5 eV. As shown in the figure, with the increase of the amount of F-doping, the area of the F 1s peak proportionately increases, reflecting an increase in the concentration of the fluorine ions in the doped material as the composition design. This observation is constant with the consecutive changes of lattice constants shown in Section 3.1, which further proves that the fluorine ions have been successfully doped into the lattice structure. | ||
| Fig. 9 The XPS spectra of the Na0.06Li0.86MnO1.98F0.02 sample and F 1s for LixMnO2−yFy. | ||
4. Conclusion
The fluorine doped monoclinic LiMnO2 could be successfully synthesized via a high-temperature solid-state reaction and subsequent ion exchange by hydrothermal method. The partial fluorine substitution with high electronegativity anions strengthens the Mn–O bands in the layered monoclinic structure, which stabilizes the structure. Though the F-doped samples had lower initial capacities, they showed better cycle performance compared with F-free samples. The Na0.05Li0.86MnO1.98F0.02 electrode delivers an initial discharge capacity of 129.2 mA h g−1 between 2 and 4.3 V at a current density of 50 mA g−1 at room temperature, which gradually increased to a maximum discharge capacity of 219 mA h g−1 after 40 cycles. The research indicated that the different performances can be attributed to the different morphologies of the respective materials as well as the effect of sodium ion concentration within the structure. Therefore, controlling the morphology of the material and the amount of sodium ions in its structure may be advantageous for developing high quality cathode materials which can be diversified for specific applications in Li-ion batteries.Acknowledgements
This work was supported by The National Natural Science Foundation of China (21061015) and The Nature Science Foundation from Xinjiang autonomous region (2015211A037).References
- J. Reed and G. Ceder, Chem. Rev., 2004, 104, 4513–4534 CrossRef CAS.
- P. Suresh, A. K. Shukla and N. Munichandraiah, Electrochem. Solid-State Lett., 2005, 8, A263–A266 CrossRef CAS PubMed.
- B. Ammundsen, J. Desilvestro, T. Groutso, D. Hassell, J. B. Metson, E. Regan, R. Steiner and P. J. Pickering, J. Electrochem. Soc., 2000, 147, 4078–4082 CrossRef CAS PubMed.
- J. E. Greedan, N. P. Raju and I. J. Davidson, J. Solid State Chem., 1997, 128, 209–214 CrossRef CAS.
- Y. J. Wei, H. Ehrenberg, N. N. Bramnik, K. Nikolowski, C. Baehtz and H. Fuess, Solid State Ionics, 2007, 178, 253–257 CrossRef CAS PubMed.
- T. J. Kim, D. Son, J. Cho and B. Park, J. Power Sources, 2006, 154, 268–272 CrossRef CAS PubMed.
- A. R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499–500 CrossRef CAS PubMed.
- F. Capitaine, P. Gravereau and C. Delmas, Solid State Ionics, 1996, 89, 197–202 CrossRef CAS.
- Z. F. Huang, X. Meng, C. Z. Wang, Y. Sun and G. Chen, J. Power Sources, 2006, 158, 1394–1400 CrossRef CAS PubMed.
- R. Prasad, R. Benedek and M. M. Thackeray, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 134111 CrossRef.
- G. Ceder and S. K. Mishra, Electrochem. Solid-State Lett., 1999, 2, 550–552 CrossRef CAS PubMed.
- C. W. Park, I. R. Mangani, H. W. Ryu, C. J. Park, J. S. Lee, S. J. Song, J. H. Moon and J. Kim, J. Phys. Chem. Solids, 2007, 68, 1126–1130 CrossRef CAS PubMed.
- A. D. Robertson, A. R. Armstrong and P. G. Bruce, Chem. Mater., 2001, 13, 2380 CrossRef CAS.
- L. H. Yu, H. X. Yang, X. P. Ai and Y. L. Cao, J. Phys. Chem. B, 2005, 109, 1148–1154 CrossRef CAS PubMed.
- B. Patoux, M. Dolle and M. M. Doeff, Chem. Mater., 2005, 17, 1044–1054 CrossRef.
- J. H. Kim, C. W. Park and Y. K. Sun, Solid State Ionics, 2003, 164, 43–49 CrossRef CAS PubMed.
- B. J. Hwang, Y. W. Tsai, C. H. Chen and R. Santhanam, J. Mater. Chem., 2003, 13, 1962–1968 RSC.
- W. K. Pang, J. Y. Lee, Y. S. Wei and S. H. Wu, Mater. Chem. Phys., 2013, 139, 241–246 CrossRef CAS PubMed.
- G. H. Kim, S. T. Myung, H. S. Kim, C. S. Yoon and Y. K. Sun, J. Electrochem. Soc., 2005, 152, A1707–A1713 CrossRef CAS PubMed.
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2013, 105, 200–208 CrossRef CAS PubMed.
- S. Jouanneau and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A1749–A1754 CrossRef CAS PubMed.
- Q. G. Zhang, T. Y. Peng, D. Zhan and X. H. Hu, J. Power Sources, 2014, 250, 40–49 CrossRef CAS PubMed.
- J. L. Xie, X. Huang, Z. B. Zhu and J. H. Dai, Ceram. Int., 2011, 37, 419–421 CrossRef CAS PubMed.
- W. S. Fyfe, Anal. Chem., 1951, 23(1), 174–175 CrossRef CAS.
- R. Chitrakar, H. Kanoh, Y. S. Kim, Y. Miyai and K. Ooi, J. Solid State Chem., 2001, 160, 69–76 CrossRef CAS.
- S. H. Kang, I. Belharouak, Y. K. Sun and K. Amine, J. Power Sources, 2005, 146, 650–653 CrossRef CAS PubMed.
- P. Barboux, J. M. Tarascon and F. K. Shokoohi, J. Solid State Chem., 1991, 94, 185–196 CrossRef CAS.
- E. Wolska, P. Piszora, W. Nowicki and J. Darul, Int. J. Inorg. Mater., 2001, 6, 503–507 CrossRef.
- L. K. Kang, M. M. Zhang, Z. H. Liu and K. Ooi, Spectrochim. Acta, Part A, 2007, 67, 864–869 CrossRef PubMed.
- H. D. B. Jenkins, H. K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef CAS PubMed.
- X. Q. Wang, H. Nakamura and M. Yoshio, J. Power Sources, 2002, 110, 19–26 CrossRef CAS.
- Y.-S. He, L. Pei, X.-Z. Liao and Z.-F. Ma, J. Fluorine Chem., 2007, 128, 139–143 CrossRef CAS PubMed.
- W. Oh, S. H. Park, J. H. Kim, Y. C. Bae and Y. K. Sun, J. Power Sources, 2006, 157, 464–470 CrossRef PubMed.
- G. H. Kim, M. H. Kim, S. T. Myung and Y. K. Sun, J. Power Sources, 2005, 146, 602–605 CrossRef CAS PubMed.
- Y. S. He, L. Pei, X. Z. Liao and Z. F. Ma, J. Fluorine Chem., 2007, 128, 139–143 CrossRef CAS PubMed.
- Y.-II Jang, B. Y. Huang, H. F. Wang, R. S. Donald and Y. M. Chiang, J. Electrochem. Soc., 1999, 146(9), 3217–3223 CrossRef CAS PubMed.
- P. Yue, Z. X. Wang, X. H. Li, X. H. Xiong, J. X. Wang, X. W. Wu and H. J. Guo, Electrochim. Acta, 2013, 95, 112–118 CrossRef CAS PubMed.
- S. Bach, J. P. Pereira-Ramos and P. Willmann, Electrochim. Acta, 2006, 52, 504–510 CrossRef CAS PubMed.
- J. M. Paulsen, C. L. Thomas and J. R. Dahn, J. Electrochem. Soc., 1999, 146, 3560–3565 CrossRef CAS PubMed.
- S. W. Mai, M. Q. Xu, X. L. Liao, L. D. Xing and W. S. Li, J. Power Sources, 2015, 273, 816–822 CrossRef CAS PubMed.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- V. I. Nefedov, Y. V. Salyn, G. Leonhsardt and R. Scheibe, J. Electron Spectrosc. Relat. Phenom., 1997, 10, 121–124 CrossRef.
- R. G. Hayes, N. Edelstein and D. A. Shirley, Electron Spectroscopy, North-Holland Publishing Company, Amsterdam, 771-781, 1972 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |